Aortopulmonary window
Images


Summary
A 16-year-old woman presented with progressive dyspnea and syncope episodes. The patient had had a personal history of a murmur since childhood, which was clinically diagnosed without additional studies, and had remained healthy until the onset of symptoms. Echocardiography revealed severe pulmonary hypertension, and chest radiography identified signs of pulmonary hypertension.
Diagnosis
Aortopulmonary window with severe nonreactive pulmonary hypertension
Findings
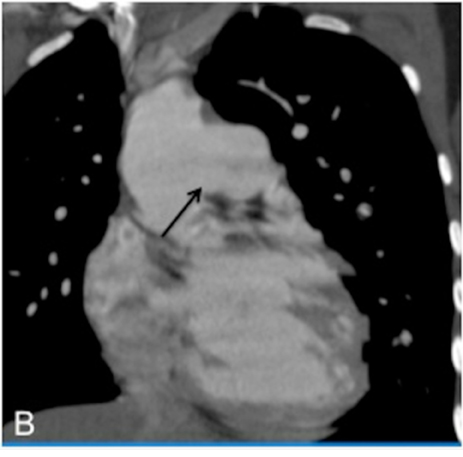
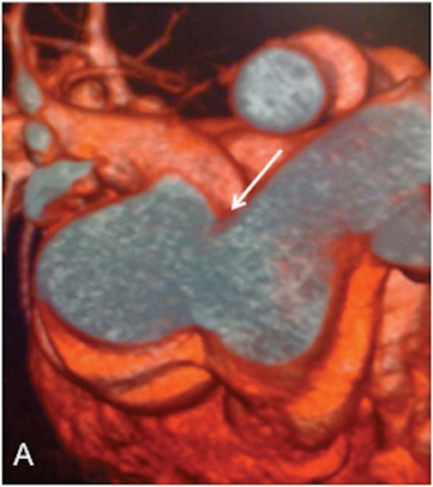
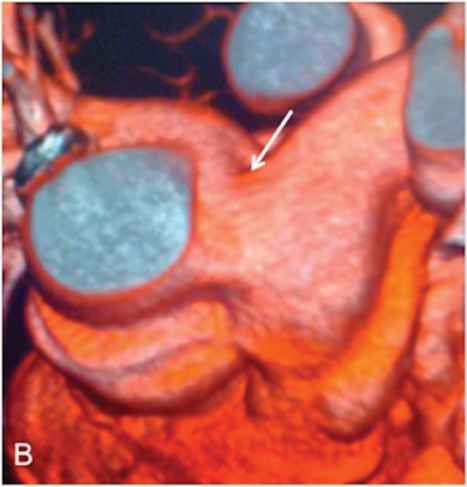
As a ligament injury was suspected to Computed tomography angiography (CTA) demonstrated a large communication between the ascending aorta and the pulmonary artery trunk (Figures 1 and 2), with a 32-mm diameter defect in the antero-posterior axis and signs of pulmonary hypertension.
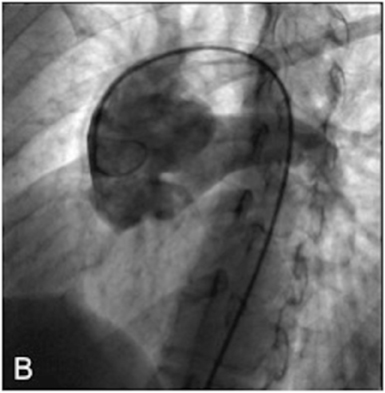
Right heart catheterization showed a significant short circuit between the ascending aorta and the pulmonary artery trunk (Figure 3)with severe pulmonary hypertension nonreactive to intravenous nitrates and 100% oxygen.
Discussion
Aortopulmonary window is caused by the failure to fuse of the two opposing conotruncal ridges that are responsible for separating the truncus arteriosus into the aorta and pulmonary artery. This defect may be found anywhere from just above the semilunar valves to the more distal ascending aorta and main pulmonary artery.1
It is a rare lesion and its prevalence among patients with congenital heart disease is only 0.1% to 0.2%. Currently, approximately 300 cases are cited in the literature1 in which aortopulmonary window occurs as an isolated lesion. It can also be associated with other cardiac abnormalities in one-third to one-half of cases. The most common associated lesions are arch abnormalities, specifically interrupted aortic arch, and coarctation of the aorta.1-2 Abnormal origin of the coronary arteries is commonly associated with aortopulmonary window.The coronary arteries may arise from the edge of the defect or on the pulmonary artery side of the defect. In addition, a variety of other lesions can occur, though rarely,with aortopulmonary window. These include ventricular septal defect (VSD), tetralogy of Fallot, and transposition of the great vessels.3
Despite the rarity of aortopulmonary window, the condition has been classified into 3 types: proximal, or type I; distal, or type II; and total, or type III. It has been suggested that, when associated with interrupted aortic arch, aortopulmonary windows are larger with greater distal extension.4
In most patients, these alterations cause a severe short circuit from left to right—similar to that of patients with left-to-right shunts,such as patent ductus arteriosus or VSD—leading to the development of congestive heart failure with dyspnea and hepatomegaly5 mainly during the first year of life or later in childhood. Pulmonary hypertension can be fatal in almost all cases if not treated early in childhood or adolescence;6 the few surviving adult patients have symptoms associated with severe pulmonary hypertension, making these cases inoperable.7 There are some reports in the literature of successful surgery in adult patients8 and some are candidates for heart-lung transplant. Physical examination demonstrates a tachypneic infant with accessory respiratory muscle use. Cardiac examination reveals an enlarged heart and, like patients with patent ductus arteriosus, the pulses are bounding. A systolic murmur can be heard along the left sternal border; however, unlike patients with a patent ductus arteriosus, a diastolic component to the murmur is rare.6 Chest x-rays reveal cardiomegaly and increased pulmonary vascular markings consistent with increased pulmonary blood flow. Diagnosis is made with echocardiography. The location and size of the communication, as well as associated anomalies, are carefully identified.
Cardiac catheterization is rarely indicated and reserved for patients who present after early infancy and, therefore, are at risk for elevated pulmonary vascular resistance, or any patient in whom the anatomy cannot be adequately defined by echocardiography.9
Multidetector CT (MDCT) and magnetic resonance imaging (MRI) can noninvasively demonstrate the defect and signs of pulmonary hypertension. Furthermore, MRI permits evaluation of the direction and quantification of the flow and the presence of other associated anomalies.10
Conclusion
Aortopulmonary window is a rare defect caused by failure of the 2 opposing conotruncal ridges to fuse, which may occur as an isolated lesion or in association with other cardiac abnormalities. Currently, early mortality following repair of simple aortopulmonary window approaches zero and depends on the presence of associated lesions. Long-term outcomes should be excellent. MDCT and MRI are useful noninvasive techniques that can could demonstrate the defect and signs of pulmonary hypertension.
- Moruno Tirado A, Santos De Soto J, Grueso Montero J, et al. Aortopulmonary window: Clinical assessment and surgical results. Rev Esp Cardiol. 2002;55:266-270.
- Soares AM, Atik E, Cortez TM, et al. Aortopulmonary window. Clinical and surgical assessment of 18 cases. Arq Bras Cardiol. 1999;73:59-74.
- Erez E, Dagan O, Georghiou GP, et al. Surgical management of aortopulmonary window and associated lesions. Ann Thorac Surg. 2004;77:484-487.
- Mori K, Ando M, Takao A, et al. Distal type of aortopulmonary window. Report of 4 cases. Br Heart J. 1978;40:681-689.
- Santos W, Rossi R, Abecasis M, et al. Aortopulmonary window--a review of nine cases. Rev Port Cardiol. 2008;27:1453-1462.
- Morrow AG, Greenfield LJ, Braunwald E. Congenital aortopulmonary septal defect. Clinical and hemodynamic findings, surgical technic, and results of operative correction. Circulation. 1962;25:463-476.
- Perloff JK. The clinical recognition of congenital heart disease. 4th ed. W.B. Saunders; Philadelphia, PA: 1994.
- Aggarwal SK, Mishra J, Sai V, et al. Aortopulmonary window in adults: Diagnosis and treatment of late-presenting patients. Congenit Heart Dis. 2008;3:341-346.
- Trehan V, Nigam A, Tyagi S. Percutaneous closure of nonrestrictive aortopulmonary window in three infants. Catheter Cardiovasc Interv. 2008;71:405-411.
- Wang ZJ, Reddy GP, Gotway MB, et al. Cardiovascular shunts: MR imaging evaluation. Radiographics. 2003;23 Spec No:S181-194.
Related Articles
Citation
Aortopulmonary window. Appl Radiol.
July 4, 2013