Tumor-induced osteomalacia (TIO) (also known as oncogenic osteomalacia)
Images
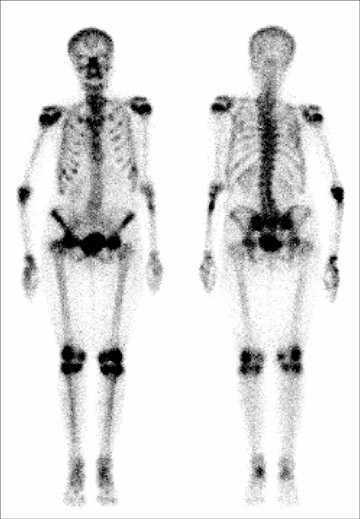
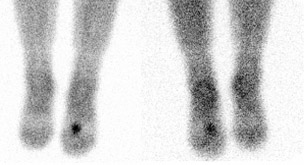
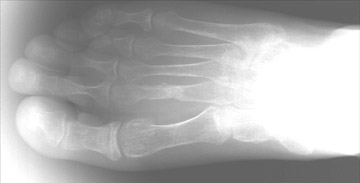

Tumor-induced osteomalacia (TIO) (also known as oncogenic osteomalacia)
CASE FOLLOW-UP
The histopathologic analysis revealed that the metatarsal lesion was a mesenchymal tumor. The resection of the tumor cured her osseous abnormalities. Soon after surgery, she recovered, resumed her normal life, and went back to jogging.
Findings
Multiple bony deformities that were suggestive of old malunited fractures that involved the left humerus, rib cage, and spine were noted on a technetium (Tc)-99m methylene diphosphate (MDP) whole-body bone scan (Figure 1). The presence of a small radiodense lesion in the left foot was detected by on an indium-111 (In-111) OctreoScan (Covidien, Hazelwood, MO), which indicated the possibility of a tumor (Figure 2). A plain X-ray of the left foot revealed the cortical destruction of the second metatarsal bone in the region of diaphysis (Figure 3). Magnetic resonace imaging (MRI) of the left foot revealed a bone-destructive tumor that involved the second metatarsal bone (Figure 4).
Discussion
Oncogenic osteomalacia is an unusual syndrome that is characterized by multiple biochemical abnormalities, such as hypophosphatemia, hyperphosphaturia, and low levels of plasma 1,25-dihydroxyvitamin D. These abnormalities produce osteomalacia in adults and rickets in children, which clinically manifest as muscle weakness, bone pain, and multiple pathologic fractures. In children, it can also lead to skeletal deformities, growth retardation, and gait problems. Stress and insufficiency fractures are prominent features. Tumors producing this syndrome secrete a substance that inhibits the renal tubular reabsorption of phosphates, which produces a cascade of biochemical abnormalities. By virtue of this property, it belongs to the family of paraneoplastic syndromes. It is interesting that all biochemical and clinical features revert to normal when the tumor is removed.1
McCance2 is credited for having described the first case in 1947, but in 1959 Prader3 and associates first recognized the relationship between the tumor and the disease in an 11-year-old girl who developed rickets in association with giant cell granuloma of a rib. All biochemical and clinical abnormalities reverted to normalcy when the tumor was removed. They postulated that the tumor produced a substance that acted like an antagonist to vitamin D or as a phosphoric agent simulating parathyroid hormone. It is now known that this unknown substance is of low molecular weight and is termed phosphatonin.4 To date, >150 cases have been described sporadically in different literatures, and, undoubtedly, many more cases have not been reported. Awareness among the clinicians and the availability of better diagnostic tools have increased the suspicion and the detection of this condition.
The majority of these tumors are located in the extremities (skin, muscles, bones) or around the head (paranasal sinuses), but they may occur in almost any part of the body. These tumors are slow growing and often remain hidden or undetected until clinical features reach a fairly advanced stage. In one review of head and neck tumors, the diagnosis of these tumors lasted a mean of 4.7 years from the onset of osteo-malacia.5 Often the clinical presentation mimics X-linked hypophosphatemia (XLH) or hereditary autosomal dominant hypophosphatemic rickets (ADHR). Excessive paraneoplastic production of phosphatonin is thought to be involved in the etiology of tumor-induced osteomalacia, whereas impaired degradation and processing of phosphatonin due to endopeptidase mutations (PHEX gene: phosphate-regulating gene with homologies to endopeptidases on the X chromosome) is thought to cause the typical findings in XLH. The fibroblast growth factor (FGF)-23 gene is associated with ADHR. Both XLH and ADHR typically present in childhood, although ADHR can present with variable or delayed age of onset.
Tumors that cause TIO are often small, slow-growing, vascular, and benign; they are associated with a variety of histologic types and are commonly mesenchymal in origin. Hemangiopericytoma is the most dominant histologic diagnosis noted in TIO. However, malignant tumors have occasionally been reported. Based on their histologic features, they have been sub-divided into 4 types: 1) phosphaturic mesenchymal tumor, mixed connective tissue type (PMTMCT); 2) osteoblastoma-like tumors; 3) ossifying fibroma-like tumors; and 4) nonossifying fibroma-like tumors.6 Hemangiopericytoma is the subtype of PMTMCT that comprises approximately 70% to 80% of all tumors associated with TIO.7 Fibroblast growth factor 23 belongs to the mesenchymal origin family, and the measurement of serum FGF-23 has been found to be of considerable importance in facilitating early diagnosis.
An important aspect of patient management is to locate the tumor. CT and/or MRI (often followed by MR angiography) are the main imaging tools used to determine tumor location. However, whole-body CT and MRI have their own limitations. Nuclear scintigraphy has emerged as a very useful and economical tool to detect and determine the site of these tumors. Once the site is identified, regional CT and/or MRI are performed to further characterize the tumor. A Tc-99m MDP bone scan has been used as an initial tool for the evaluation of the skeletal system. Diffuse skeletal uptake often presents as a “superscan” with focal up-take at sites of fractures. Multifocal lesions of increased activity seen in skeletal scintigraphy of patients with acquired hypophosphatemic osteomalacia may be misinterpreted as osseous metastases. Gallium-67 is useful for imaging areas of infection, inflammation, and tumors, including those tumors that induce osteomalacia. Gallium scanning to determine the staging of tumor-induced osteomalacia has also been described. In 1 reported case, a knee tumor that was responsible for producing TIO was detected by whole-body Tl-201 and (metaiodobenzylguanidine) Tc-99m scintigraphy.8 Recently, whole-body Tc-99m methoxyisobutyl-isonitrile (MIBI) scanning has been described as a part of simplified protocol that is cost- and time-effective. The authors recommended that whole-body MRI scans should be reserved only for patients with Tc-99m MIBI-negative scans.9
Many of these tumors express somatostatin receptors that regulate secretory activity, as has been shown by other similar neuroendocrine tumors. Octreotide-labeled In-111 scanning has been successfully used for the detection of such tumors,10 as is also seen in the present case. On the basis of an octreotide scan, TIO can be classified as OctreoScan-positive tumors or OctreoScan-negative tumors. This classification also has bearing on the treatment. OctreoScan-negative tumors should be further evaluated by cost- effective nuclear scans followed by limited MRI. The role of [18]-fluorodeoxyglucose positron emission tomography (PET)/CT imaging is well established in the detection of various tumors, and its role in TIO is now being defined, with encouraging results. However, PET scans usually do not include the limbs. It is mandatory to include the limbs in the field, as these tumors are common in the limbs and may be easily missed.
CONCLUSION
Unexplained generalized bone pain or multiple fractures must be tested for calcium and phosphate homeostasis for a complete diagnostic work-up. Detection of renal phosphate wasting indicates the need for further evaluation for possible hereditary and acquired causes.
On the basis of the OctreoScan results, cases of TIO should be classified as either OctreoScan-positive tumors or OctreoScan-negative tumors. This classification also has a bearing on the treatment. OctreoScan-negative tumors should be further evaluated with cost-effective nuclear scans. Once the tumor is detected, limited MRI should be performed. For PET scan evaluation, it is mandatory to include the limbs in the field (a vertex-to-toes protocol), as these tumors are common in the limbs and may easily be missed if the limbs are truncated.
- Fanconi A, Fischer JA, Prader A. Serum parathyroid hormone concentrations in hypophosphatemic vitamin D resistant rickets. Helv Paediatr Act. 1974;29:187-194.
- McCance RA. Osteomalacia with Looser's nodes (Milkman's syndrome) due to a raised resistance to vitamin D acquired about the age of 15 years. Q J Med. 1947;16:33-
- Prader A, Illing R, Uehlinger E, Stalder G. Rickets following bone tumor. Rachitis infolge knochentumors. [In German.] Helv Paediatr Acta. 1959;14:554-565.
- Econs MJ, Drezner MK. Tumor-induced osteomalacia-unveiling a new hormone. New Eng J Med. 1994;330:1679-1681.
- Gonzalez-Compta X, Manos-Pujol M, Folia-Fernandez M, et al. Oncogenic osteomalacia: Case report and review of head and neck associated tumors. J Laryngol Otol. 1998;112:389-392.
- Weidner N, Santa Cruz D. Phosphaturic mesenchymal tumors. A polymorphous group causing osteomalacia or rickets. Cancer. 1987;59:1442-1454.
- Folpe AL, Fanburg-Smith JC, Billings SD, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: An analysis of 32 cases and a comprehensive review of the literature. Am J Surg Path. 2004;28:1-30.
- Kimizuka T, Ozaki Y, Sumi Y. Usefulness of 201Tl and 99mTc MIBI scintigraphy in a case of oncogenic osteomalacia. Ann Nucl Med. 2004;18:63-67.
- Gambhir S, Kongara S, Pawaskar A, et al. Simplified protocol for detection and localization of tumor causing adult onset sporadic hypophosphatemic osteomalacia. Abstract No.1039. J Nuc Med. 2005;46(2 Abstract Book):317-318.
- Rhee Y, Lee JD, Shin KH et al. Oncogenic osteomalacia associated with mesenchymal tumor detected by indium-111 octreotide scientigraphy. Clin Endocrinol (Oxf). 2001;54:551-554.