Navajo Neurohepatopathy
A Native American infant initially presented with increased abdominal girth and a tense abdomen. Laboratory abnormalities included elevated bilirubin. An ultrasound and liver biopsy were performed. Pathology noted decreased mitochondrial quantity, large and numerous lysosomal granules, microvesicular steatosis, and bile canaliculi distention. The patient was referred for genetic testing.
Case Summary
A Native American infant initially presented with increased abdominal girth and a tense abdomen. Laboratory abnormalities included elevated bilirubin. An ultrasound and liver biopsy were performed. Pathology noted decreased mitochondrial quantity, large and numerous lysosomal granules, microvesicular steatosis, and bile canaliculi distention. The patient was referred for genetic testing.
In childhood, the patient represented with new-onset ascites. Electroencephalography (EEG) and brain MRI were performed due to agitation and a decline in mental status.
Imaging Findings
The patient underwent serial ultrasound examinations. The initial scans were normal, while the last demonstrated a cirrhotic liver with multiple nodules and ascites. A subsequent MRI examination of the abdomen with liver elastography demonstrated numerous subcentimeter-sized hepatic nodules, most compatible with regenerative nodules, and increased mean liver stiffness (6.03 kPa), consistent with cirrhosis (in children aged 3–5 years, the mean is ~3.7, and the 75th percentile is ~3.9).1
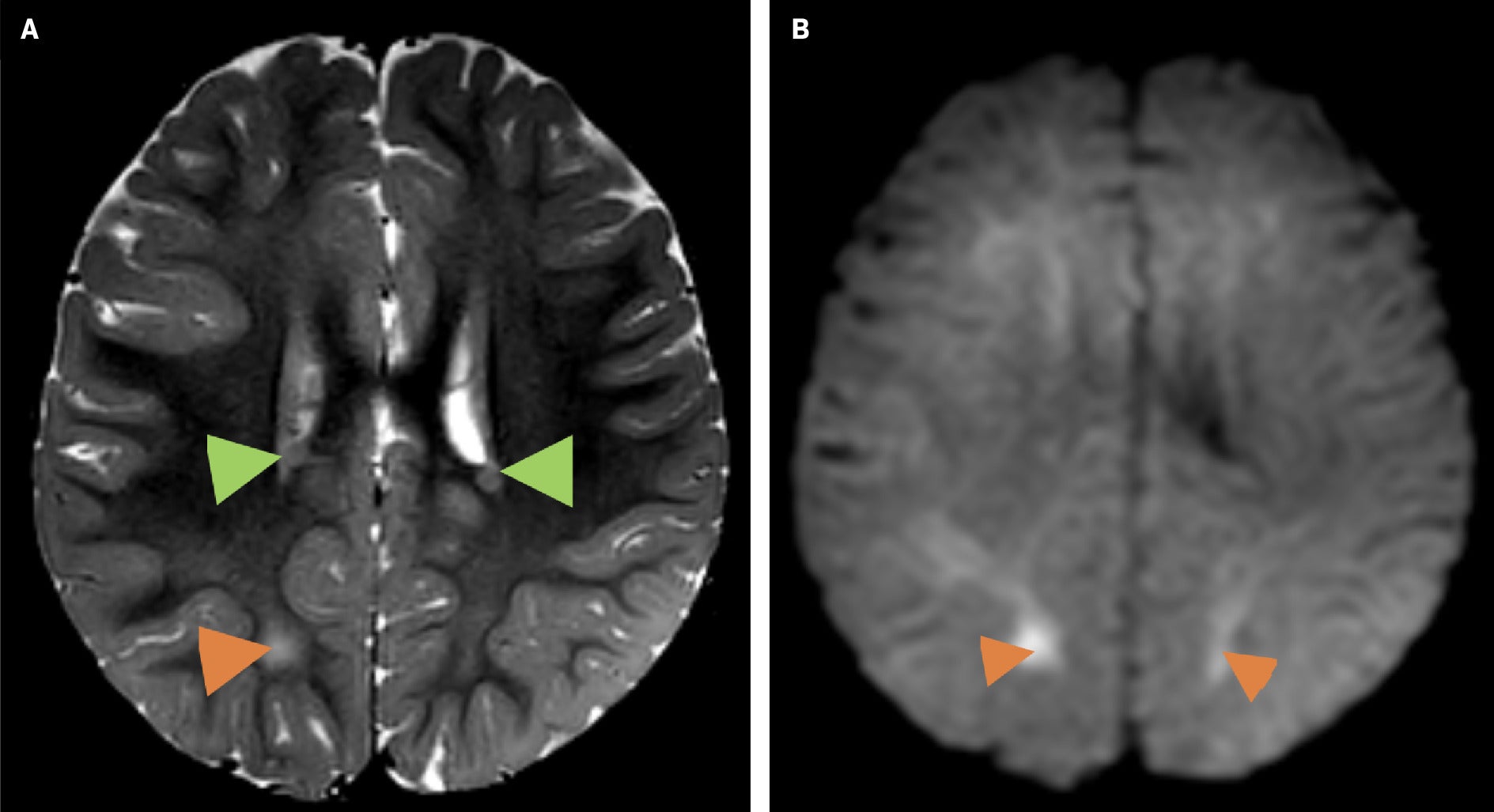
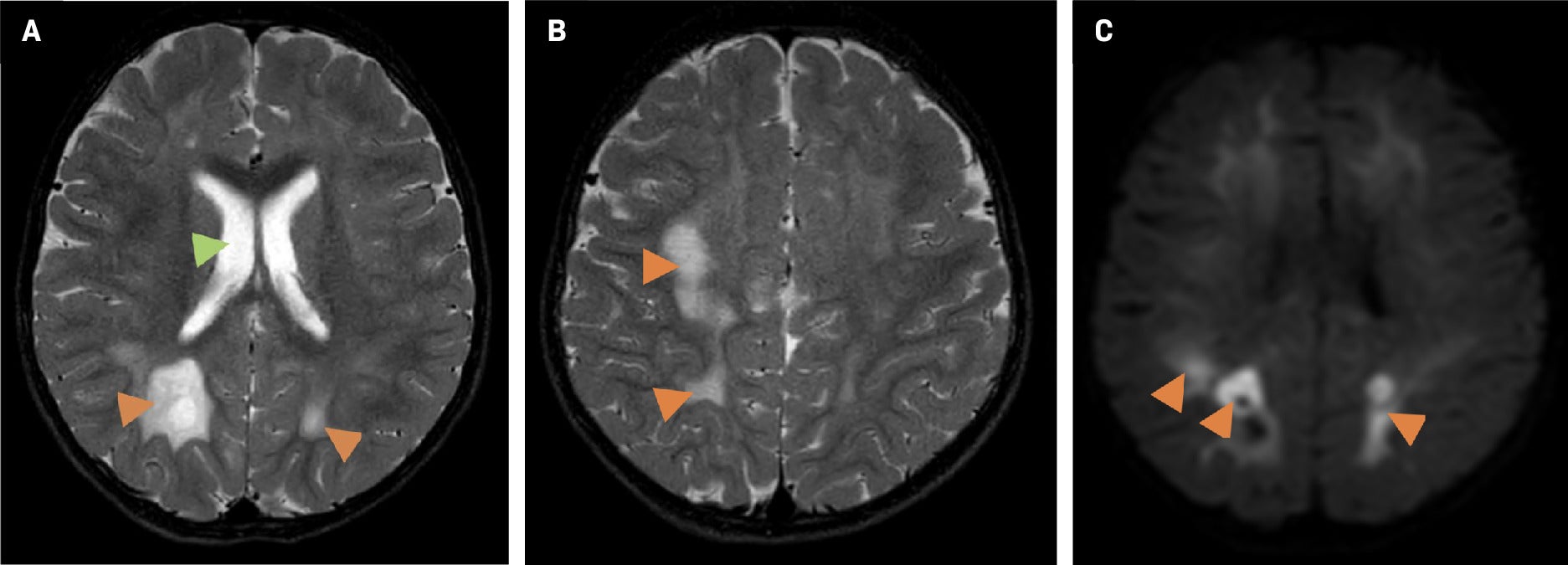
Altered mental status prompted an EEG and brain MRI. The EEG showed left hemispheric abnormalities, including continuous diffuse polymorphic delta slowing and continuous mild voltage attenuation. The MRI demonstrated bilateral white matter T2 hyperintensity, diffusion restriction, and bilateral multifocal periventricular heterotopic gray matter of unknown significance, although gray matter heterotopia is associated with a wide range of neurodevelopmental disorders ( Figure 1 ). A follow-up brain MRI several months later demonstrated a worsening abnormal white matter signal throughout the supratentorial brain and cerebellum with associated diffuse restriction, as well as slight ventricular enlargement commensurate with the degree of parenchymal atrophy ( Figure 2 ).
Brain MRI. Axial T2 (A) showing bilateral white matter T2 hyperintensities (orange arrowheads) and bilateral periventricular heterotopic gray matter foci (green arrowheads), and axial diffusion-weighted imaging (DWI) (B) showing foci of diffusion restriction (orange arrowheads).
Brain MRI one year later. Axial T2 (A, B) demonstrates progressive abnormal white matter signal hyperintensity (orange arrowheads) and an interval ventricular enlargement (A, green arrow). Axial DWI (C) shows worsening restricted diffusion (orange arrowheads).
Diagnosis
Navajo neurohepatopathy.
Differential diagnoses that may be considered include idiopathic neonatal hepatitis, biliary atresia, and metabolic liver diseases caused by enzyme defects, such as tyrosinemia, galactosemia, Wilson’s disease, glycogen storage disorders, and lysosomal storage disorders, among others.
Discussion
Navajo neurohepatopathy (NNH) is a rare autosomal recessive mtDNA depletion syndrome (MDS) presenting in children of Navajo descent and primarily affecting the liver and nervous system.2, 3 The estimated incidence of the disease in the western Navajo Reservation is 1 in 1,600 live births.4 The cause of NNH is an R50Q mutation of the nuclear MPV17 gene that encodes an inner mitochondrial membrane protein thought to participate in mtDNA maintenance, which is necessary for normal cellular energy production.2
The average age at onset for NNH is 13 months; patients often do not survive past the first decade of life.5 Clinical features include liver disease, sensory and motor neuropathy, failure to thrive, corneal anesthesia and scarring, central nervous system demyelination, and recurrent metabolic acidosis, along with other concurrent illnesses.4, 5 Based on the diagnostic criteria of Holve et al,4 NNH may be diagnosed with confidence if four of the criteria listed above are present or if three are present and the patient has an affected sibling.
As clinical diagnosis is difficult prior to disease progression, imaging studies are a key tool in identification. Liver imaging and biopsy may offer a more timely diagnosis. However, the use of biopsy may be limited owing to uncommon knowledge of this rare disease among pathologists. Therefore, genetic testing offers the most definitive and potentially earliest diagnosis.
Typical MRI brain imaging shows diffuse leukoencephalopathy, manifesting as an abnormal, hyperintense T2 signal throughout the cerebral white matter, along with areas of restricted diffusion.2, 4 This differs from other mitochondrial disorders that predominantly affect the cortex and deep gray nuclei. Currently, there is no curative treatment for NNH.6, 7 The only treatment option for liver disease is transplantation;8 however, this is only palliative and does not alter long-term neurological decline.
This case demonstrates the difficulty of diagnosing NNH through imaging studies and liver biopsy alone. Suspicion of NNH should be high in pediatric patients of Navajo descent presenting with liver pathology and should prompt genetic testing for the disease as well as brain MRI.
Conclusion
Navajo neurohepatopathy is a rare and incurable mitochondrial disease affecting children of Navajo descent, causing liver disease and diffuse leukoencephalopathy. Health care professionals working near and serving Indigenous populations should be familiar with the signs and symptoms of NNH to enable prompt diagnosis and supportive treatment.
References
Citation
KA S, K H, Kuwabara.Navajo Neurohepatopathy. Appl Radiol. 2025; (2):42-44.
doi:10.37549/AR-D-2025-00071
April 1, 2025