Kabuki Syndrome
Kabuki syndrome is a rare congenital disorder with a predilection for affecting the skeletal, gastrointestinal, and cardiovascular systems, as well as causing intellectual disability and developmental delay. Key imaging findings include skeletal findings such as craniofacial and inner ear abnormalities. These radiological findings can be further complemented with physical examination and genetic testing in establishing a diagnosis. Although there is no cure, treatment is focused on addressing the quality of life for patients, including symptomatic management.
Keywords: genetic syndrome, congenital heart disease, congenital eye anomalies
Clinical Summary
A male baby with Kabuki syndrome (KS) type 1 with associated congenital heart disease (DORV, tetralogy of Fallot, hypoplastic mitral valve, and ventricular septal defect), failed hearing screen, right microphthalmia with retinal detachment, left anterior staphyloma, neonatal hypoglycemia, and mild-to-moderate hypotonia was imaged to evaluate for underlying abnormalities.
Imaging
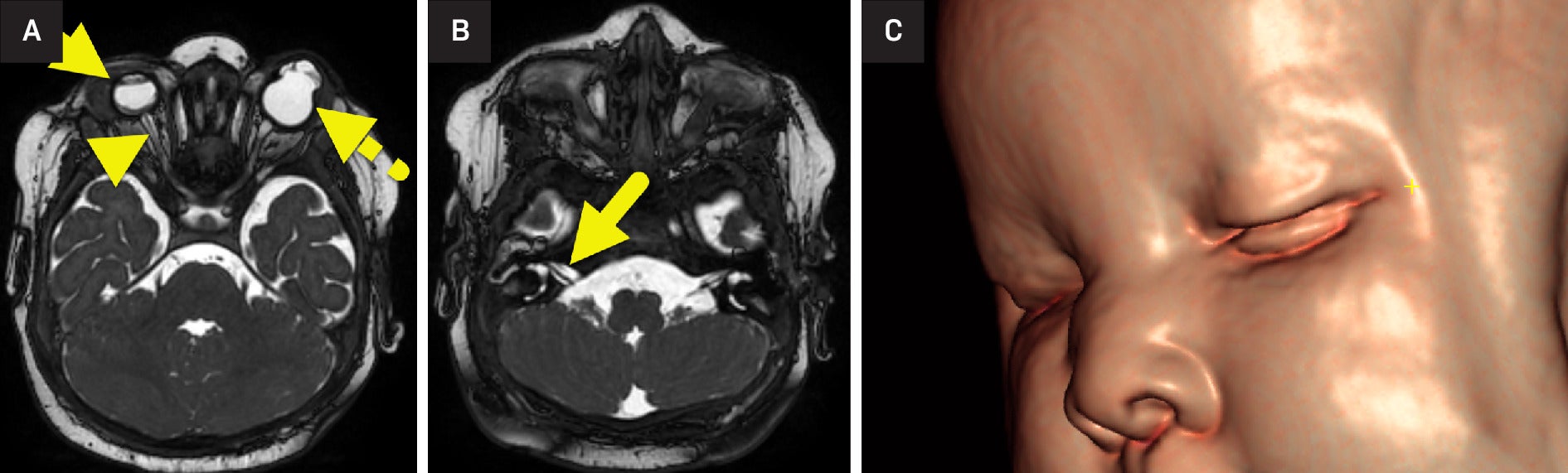
MRI of the brain and orbits ( Figure 1 ) and abdomen ( Figure 2 ) was performed. The right globe was small with a small anterior chamber and a lack of the normal lens. The retina was detached with a small vitreous hemorrhage. The right optic nerve was extremely small and asymmetric compared with the left. The left globe had a bilobed appearance with a cystic area extending into the region of the anterior chamber from the rudimentary orbit. A normal lens was not seen. The cochlear nerves were small, and the cochleae were incompletely partitioned.
(A) MRI of the orbits shows right microphthalmia (arrow). The anterior chamber of the right globe is small, and the lens is absent. The right retina is detached, and the right optic nerve (arrowhead) is small. The left globe (dashed arrow) had a bilobed appearance with a cystic area extending in the region of the anterior chamber from the rudimentary orbit. (B) Images through the internal auditory canal show small cochlear nerves (arrow). (C) 3D reconstruction of the face shows a depressed nasal bridge.
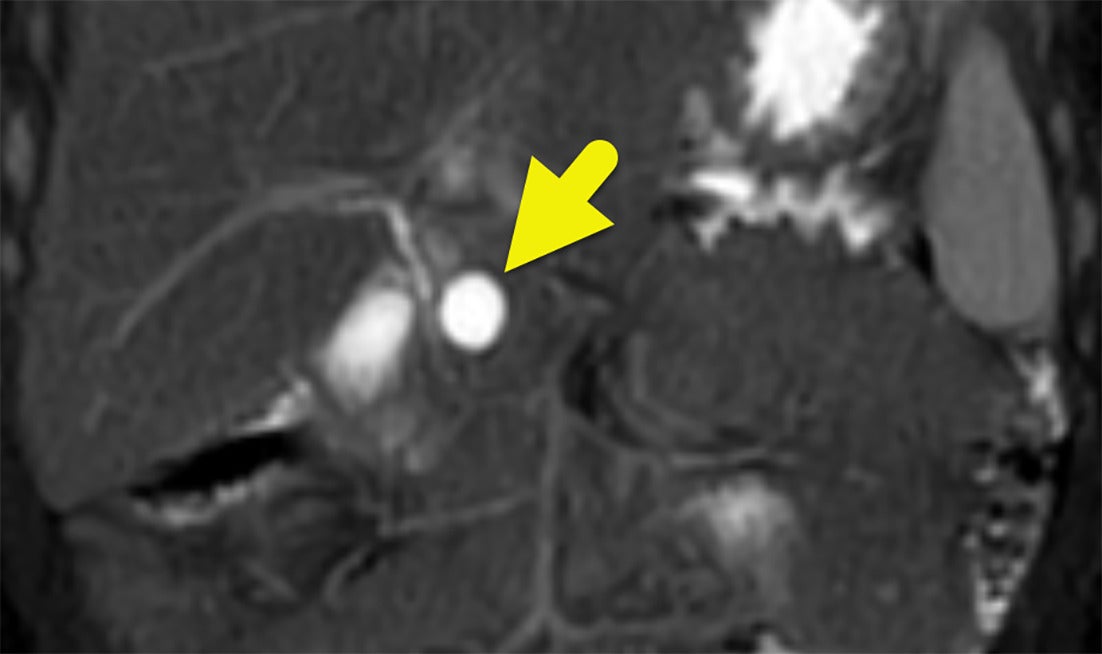
Abdominal MRI shows a cyst (arrow) in the pancreatic head. The cyst did not communicate with the common bile duct. A second cyst was present in the pancreatic tail (not shown).
Diagnosis
Kabuki syndrome.
Differential diagnosis includes CHARGE, 3MC, and Hardikar syndromes, KAT6B -related disorders, and other genetic disorders involving chromatin regulation.
Discussion
Kabuki syndrome is a rare congenital disorder that affects various parts of the body. It was previously named “Kabuki makeup syndrome” by Japanese researchers in 1981 and is characterized by distinctive facial features resembling traditional Japanese theater makeup. KS impacts craniofacial and vertebral development, as well as the cardiac, renal, and gastrointestinal systems.1 In the United States, KS affects about 1/32,000 to 1/86,000 people.2 Genetic mutations in the KMT2D and KDM6A genes have been identified to have a close association with KS. These genes serve as epigenetic modulators that regulate the activation and suppression of chromatin, which plays a vital role in DNA storage and expression.3 Spontaneous or de novo mutations in the KMT2D gene are responsible for up to 75% of known KS cases.1 Mutations in the KDM6A gene cause up to 5% of other cases, while the remaining 20% of patients do not have an identified mutation.1
Several facial characteristics support the clinical diagnosis of KS, including eversion of the lateral lower eyelids, elongated palpebral fissures, hypertelorism, long eyelashes, and arched eyebrows with thinning in the lateral third, a depressed nasal tip, and a short columella that contribute to the disease’s namesake.3, 4 Other findings include large ears, inner ear abnormalities, cleft lip or palate, and thin upper lips (3,4). Kabuki syndrome is associated with developmental delay, short stature, and intellectual disabilities.3
Patients are typically medically evaluated because of their distinctive facial features. Other features that tend to prompt further workup include feeding difficulties, hypotonia, short digits, and joint laxity. In rare instances, patients with the KDM6A mutation may present with hyperinsulinemic hypoglycemia requiring emergency diagnosis and treatment.5 Diagnosis of KS may be delayed, as certain facial features may not become evident until later in development. Patients with KS have a 30-55% chance of concurrent cardiac anomalies such as coarctation of the aorta and atrial and ventricular septal defects. Other systems may be affected, including the skeletal and renal systems.6
Common radiologic findings in KS revolve around skeletal abnormalities, such as scoliosis, hip dysplasia, clavicular pseudarthrosis, vertebral abnormalities, clubfoot, tarsal coalition, and hip and patella dislocation. Hip dislocation is found in 18-62% of individuals. Early diagnosis of these anomalies can lead to interventions, increasing the chances of successfully correcting skeletal abnormalities.7 Neuroimaging findings include a decrease in brain gray matter volume and cerebral blood flow in the precentral and frontal gyri.8
Imaging can help to clarify ambiguous or overlapping clinical presentations between KS and other similar conditions.9 Like KS, CHARGE syndrome also features distinct facies and skeletal abnormalities. Inner ear abnormalities such as absent cochlea, dilated, dysplastic vestibule, and Mondini dysplasia are found in KS but not CHARGE, while esophageal atresia is a component of CHARGE but is not present in KS (7, 8).9, 10 Patients with 3MC syndrome also have characteristic facies with hypertelorism, highly arched eyebrows, and a cleft lip or palate. However, compared with KS, 3MC syndrome is more often associated with craniosynostosis, radioulnar fusion, and caudal appendage.11 Hardikar syndrome, an extremely rare congenital disorder, has been speculated to overlap with KS. Abdominal imaging may reveal Hardikar syndrome’s higher association with GI abnormalities, such as biliary obstruction or intestinal malrotation.12
There is no cure for KS, and the management of the disorder focuses on prioritizing the quality of life for each patient. Clinical, genomic, and radiologic findings contribute valuable information to a patient’s diagnosis of KS. Speech and physical therapy can greatly benefit a patient’s long-term development. Ultimately, the management of KS requires a multidisciplinary approach with collaboration between providers to optimize patient outcomes. Depending upon the mix of abnormalities and their severity, individuals with KS can live a normal life span and participate in activities.
Conclusion
KS is a rare congenital disorder with a predilection for affecting the skeletal, gastrointestinal, and cardiovascular systems, as well as causing intellectual disability and developmental delay. Key imaging findings include skeletal findings such as craniofacial and inner ear abnormalities. These radiological findings can be further complemented with physical examination and genetic testing in establishing a diagnosis. Although there is no cure, treatment is focused on addressing the quality of life for patients, including symptomatic management.
References
Citation
Kondapalli K, Towbin RB, Morgan D, Schaefer CM, Towbin AJ.Kabuki Syndrome. Appl Radiol. 2025; (1):
doi:10.37549/JPCR-25-0001
October 1, 2025