Update on adult renal cystic diseases
Images
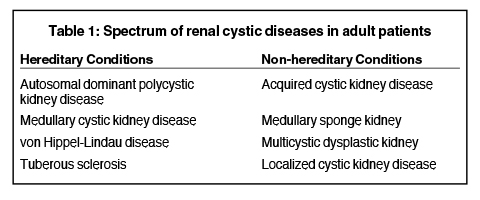
A wide spectrum of hereditary and non-hereditary disorders results in development of renal cysts in adult patients. Hereditary renal cystic syndromes include autosomal dominant polycystic kidney disease, medullary cystic kidney disease, von Hippel-Lindau syndrome, and tuberous sclerosis. The non-hereditary disorders that predispose to the formation of renal cysts include acquired cystic kidney disease, medullary sponge kidney, multicystic dysplastic kidney, and localized renal cystic disease (Table 1). 1 Recent advances in genetics and molecular biology have shown that functional and structural abnormality of the primary cilia of the renal tubular epithelial cells subsequently lead to hereditary renal cystic diseases. Non-hereditary renal cystic diseases result from abnormal renal embryogenesis and/or neoplastic mechanisms. Many adult renal cystic diseases show characteristic imaging findings that may help in accurate diagnosis. Improved understanding of pathophysiologic mechanisms has led to development and testing of novel drugs for some of these diseases. In addition to diagnosis, imaging studies play a pivotal role in long-term surveillance, assessing prognosis, and testing efficacy of drugs.
Hereditary renal cystic diseases
Autosomal dominant polycystic kidney disease
Autosomal dominant polycystic kidney disease (ADPKD), the most common inherited renal cystic disease, is characterized by marked enlargement of bilateral kidneys and multiple expansile cysts.2,3 ADPKD is a heterogenetic disorder caused by mutations in PKD1 (located at chromosome 16p13 and encodes for polycystin-1 protein) and PKD2 genes (located at 4q21 and encodes for polycystin-2 protein). Abnormality of PKD1 gene is seen in 85% of ADPKD patients and that of PKD2 gene results in the remainder.1,4 The polycystin-1 and polycystin-2 proteins are located within the primary cilia of renal tubular epithelial cells and play a major role in renal tubular homeostasis and epithelial proliferation.5,6 Disruption of polycystin signaling in ADPKD causes inability of tubular epithelial cells to sense mechanical cues that normally regulate tissue morphogenesis and result in tubular ectasia due to overgrowth of tubular epithelial cells and uncontrolled cyst formation due to increased intratubular fluid secretion.4,7,8
Imaging studies not only help in the diagnosis but also play an important role in the assessment of complications and long-term follow-up.9,10 At CT/MRI, the presence of bilateral enlarged kidneys containing innumerable simple and hemorrhagic/proteinaceous cysts is typical of ADPKD (Figure 1).1 Extra-renal manifestations of ADPKD include hepatic, pancreatic, splenic, and seminal vesicle cysts, colonic diverticula, aortic and intra-cranial arterial aneurysms, and abdominal wall hernias. About 45 % of patients will develop end-stage renal disease by the age of 60 years. Novel drugs such as vasopressin receptor blockers and mammalian target of rapamycin (mTOR) inhibitors (for example, sirolimus) are shown to decrease the rate of cyst growth and halt disease progression11. MR imaging based renal cyst volumetry is an important tool in monitoring disease progression and assessing treatment response before renal function begins to decline.9,12,13 Continued increase in total kidney and cyst volume indicates progression of disease. There is no evidence of increased risk of renal cell carcinoma in patients with ADPKD.14
Medullary cystic kidney disease
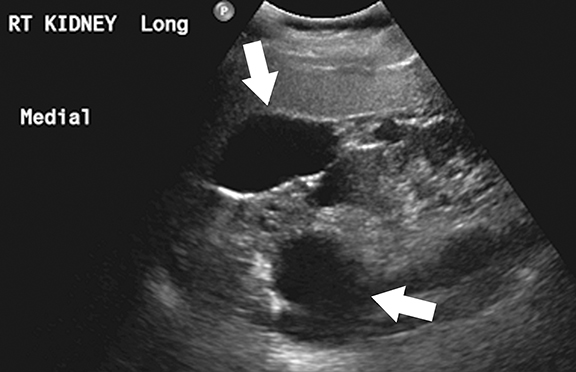
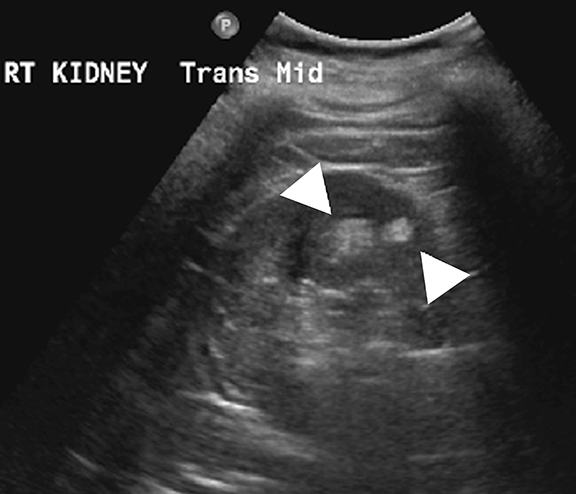
Medullary cystic kidney disease (MCKD) is a chronic, progressive kidney disease characterized by bilateral, multiple medullary cysts and tubulo-interstitial nephropathy in normal to small-sized kidneys.15 While MCKD type 1 is caused by mutations of the MCKD1 gene (located at chromosome 1q21), type 2 disease is caused by mutations of the MCKD2 gene (located on chromosome 16p12 that encodes for the uromodulin protein). 1,16 Uromodulin protein plays an important role in the formation of normal adhesion signaling complexes in renal tubules. Abnormal function due to gene mutations leads to ciliary dysfunction, which in turn causes medullary cysts.16,17 Clinically, MCKD patients present with polyuria, polydipsia, hyperuricemia, and slowly progressive renal parenchymal disease leading to end-stage renal disease.18,19 Imaging studies play an adjuvant role in the diagnosis of MCKD as family history, clinical features, and laboratory findings are the mainstay of diagnosis.1,20 Although the presence of medullary cysts supports the diagnosis of MCKD, this is not essential. At imaging, the kidneys are small to normal sized and show bilateral, multiple cysts of varying sizes at the medulla or cortico-medullary junction in bilaterally (Figure 2). 21 Renal transplantation is the definitive treatment and the cysts are not known to recur in transplanted kidneys.1,22
von Hippel-Lindau disease
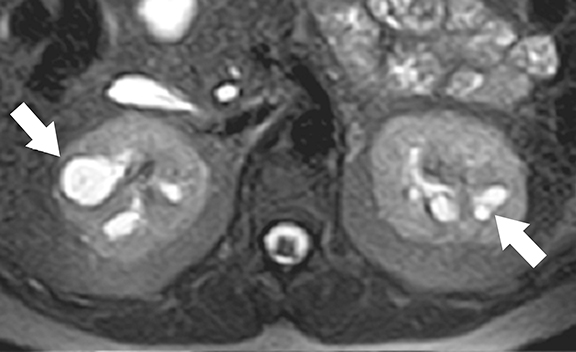
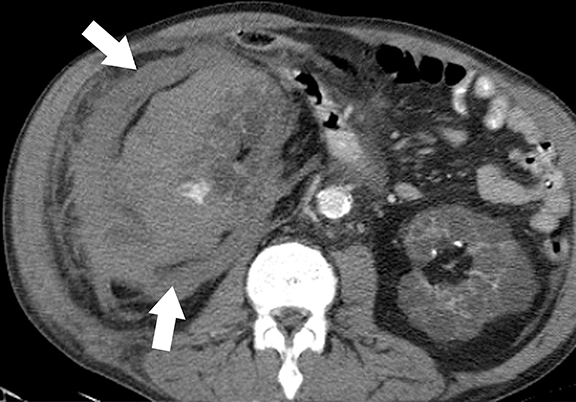
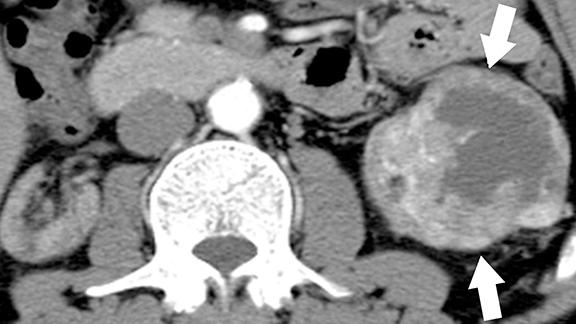
von Hippel-Lindau (VHL) disease is an autosomal dominant, inherited multisystem tumor syndrome characterized by the development of benign and malignant neoplasms of multiple organs. The disease results from inactivating mutations of the VHL tumor-suppressor gene (located at chromosome 3p25 and encoding pVHL protein).23,24 The main function of the pVHL is proteosomal degradation of hypoxia inducible factor (HIF), which is involved in homeostasis and oxygen sensing mechanism. Uninhibited up-regulation of HIF and downstream effectors in VHL patients results in uncontrolled cell proliferation and marked angiogenesis resulting in the development of hypervascular tumors.25 In addition, pVHL plays a seminal role in renal ciliogenesis, ciliary assembly, and function; inactivation of this protein causes deranged ciliary functions that cause abnormal proliferation of renal epithelium.26,27 Up to 65% of VHL patients may develop renal manifestations, which include simple and complex cysts and multiple, bilateral clear cell renal cell carcinomas.25 Common extra-renal tumors include central nervous system hemangioblastomas, retinal angiomas, pancreatic cysts and tumors, and pheochromocytomas.25 The age of onset of RCCs in VHL patients is much earlier than in sporadic cases of RCCs.28
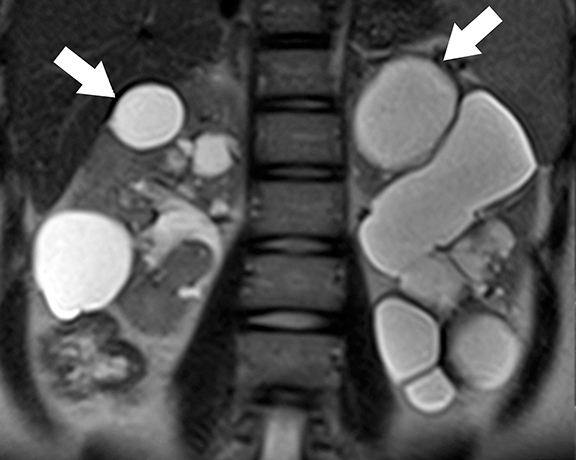
At imaging, the presence of multiple simple and complex renal cysts interspersed with complex cystic and solid enhancing renal masses is characteristic of VHL (Figure 3). 25 Multiple pancreatic cysts are commonly identified in all patients with renal findings. Although some cysts undergo involution overtime, others may grow continuously. Management options for RCCs include continued surveillance and nephron-sparing procedures such as partial nephrectomy or image-guided radiofrequency/cryo ablation. While RCCs measuring more than 3 cm in maximal diameter are usually treated, patients with smaller tumors typically undergo periodic imaging surveillance.29
Tuberous sclerosis complex
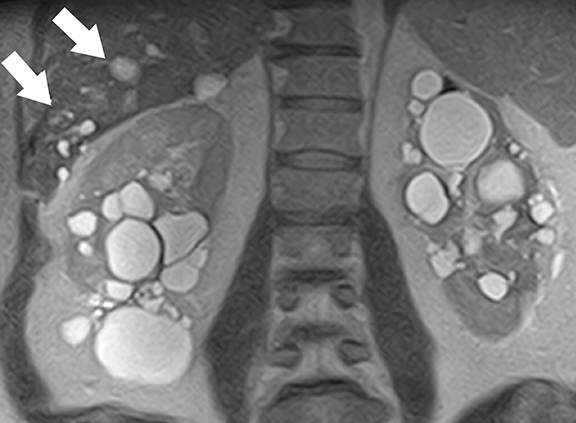
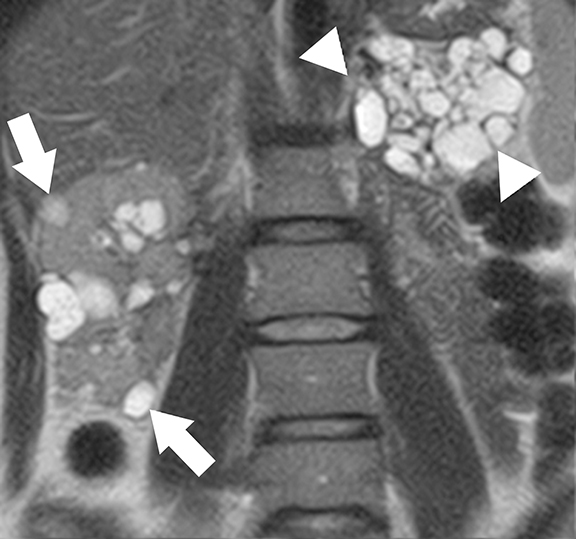
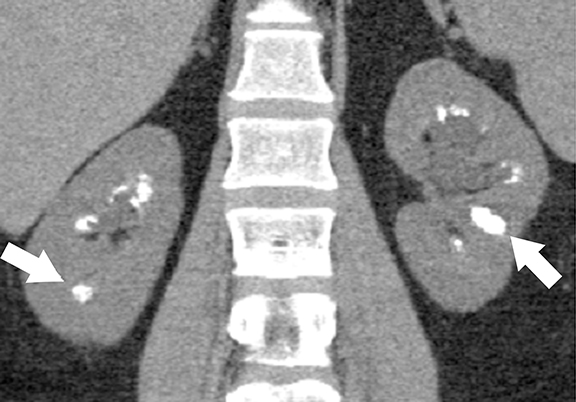
Tuberous sclerosis complex (TSC) is an autosomal dominant condition characterized by both renal and extra-renal manifestations.30 Renal involvement is seen in about 50-60% of patients, which include renal cysts (14-32%) and angiomyolipomas (30-40%). 31 Central nervous system lesions including multiple cortical tubers, subependymal nodules, subependymal giant cell astrocytomas, and white matter abnormalities are the common extra-renal manifestations in TSC patients. Inactivating mutations of TSC1 gene (located at chromosome 9q34 that encodes hamartin) and/or TSC2 genes (located at chromosome 16p13 that encodes hamartin)30-32 are responsible for the development of TSC. Tuberin-hamartin protein complex plays an important role in controlling cell growth and proliferation and functions as a tumor suppressor by suppressing mammalian target of rapamycin (mTOR); dysfunction of this protein results in ciliary dysfunction and uncontrolled activation of mTOR leading to the formation of angiomyolipomas and renal cysts.33,34
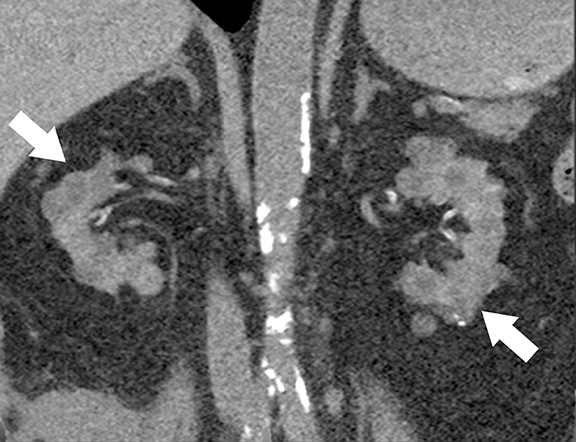
At cross-sectional imaging, multiple renal cysts intermixed with angiomyolipomas involving bilateral kidneys is the typical appearance of TSC on cross-sectional imaging (Figures 4 and 5).1,35 Interestingly, some patients may develop only simple cysts while others may have AMLs only; however, both lesions may manifest in childhood and increase in size and number as patients grow older. There is no increased risk of malignancy per se in TSC patients; however, RCC occur at a younger age in comparison to general population.35
Non-hereditary cystic kidney diseases
Acquired cystic kidney disease
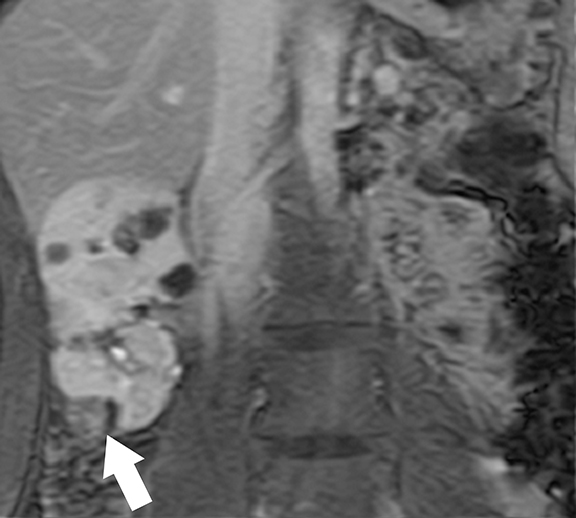
Acquired cystic kidney disease (ACKD) is characterized by the development of bilateral, multiple renal cysts (three or more per kidney) in patients with end-stage renal disease (ESRD) without any known hereditary cystic renal disease.36 Although ACKD is strongly associated with long-term hemodialysis, at least 8-13% of patients develop cysts before the initiation of dialysis.36 Compensatory hypertrophy of the functioning nephrons in ESRD patients is the initial event in ACKD; this leads to stimulation of various mitogenic factors that cause hyperplasia of renal tubular epithelium, increased fluid production resulting in multiple renal cyst formation. 37 Additionally, uncontrolled activation of growth factors and proto-oncogenes result in the development of renal cell carcinoma.14,38
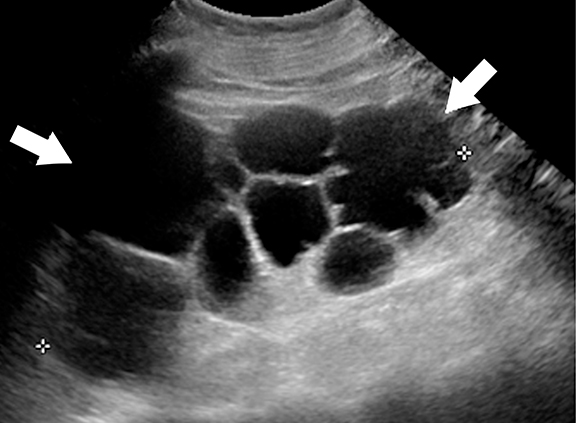
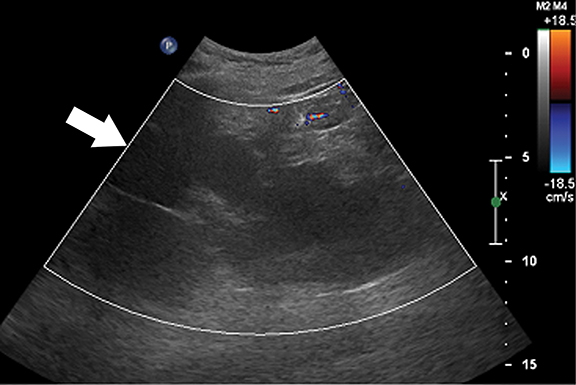
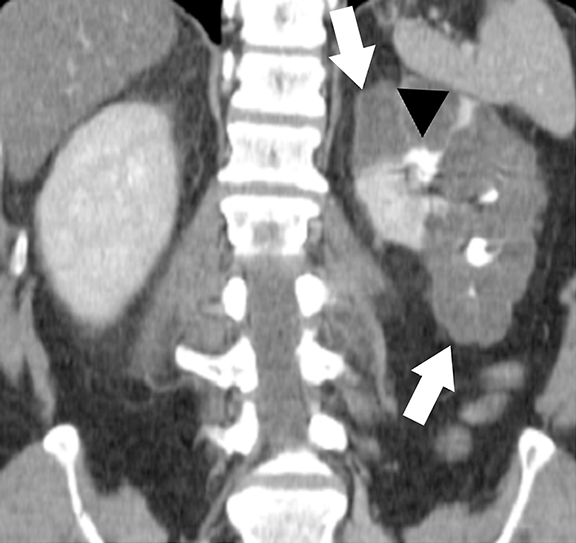
Ultrasound is the primary modality in the diagnosis of ACKD and it typically demonstrates small echogenic kidneys with multiple cysts bilaterally. At CT/MRI, bilateral small-sized kidneys with multiple cysts of varying sizes are the characteristic appearance of ACKD (Figure 6).1,36 Complications of ACKD include hemorrhage into the cysts, spontaneous cyst rupture resulting in perinephric hematoma, cyst infection, ureteral stones and development of renal cell carcinoma (up to 7% of patients) (Figure 7).36,37 ACKD-associated RCC and clear cell papillary RCC associated with ESRD are the two common histologic subtypes of RCCs; among them, ACKD-associated RCC is the most common and most aggressive form (Figure 8).39 Mostly RCCs develop within or around a preexisting cystic lesion, thus making regular US follow-up of these lesions is very important.36 Although cysts in native kidneys regress in time after renal transplantation, solid neoplasms may show a more aggressive course secondary to immunosuppression.36
Medullary sponge kidney
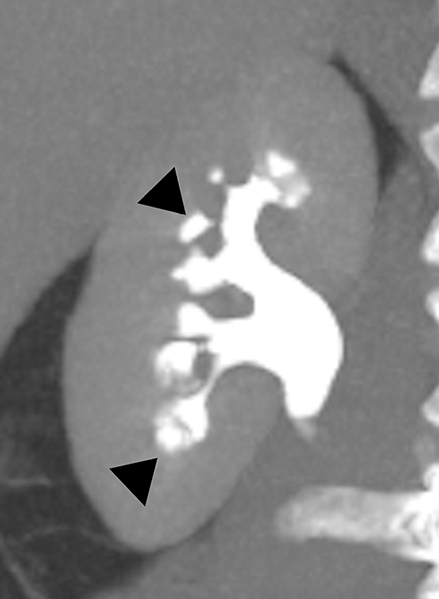
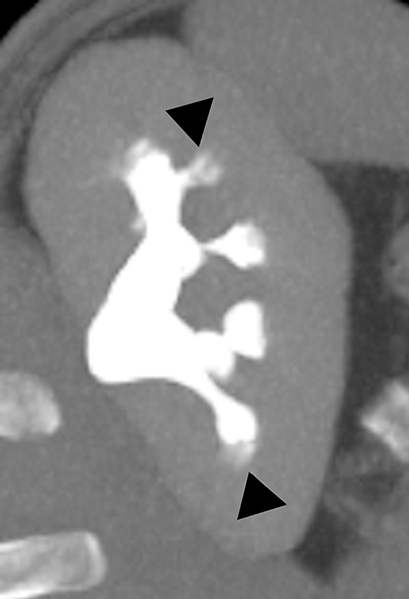
Medullary sponge kidney (MSK) is a developmental anomaly that results from diffuse or focal ectasia and cystic dilatation of the collecting ducts in the renal pyramids.40 It is hypothesized to result from disruption of the interface between the developing ureteral bud and metanephric blastema during embryogenesis.40 Most of the patients are asymptomatic; renal colic, hematuria, recurrent urinary tract infections are common presenting symptoms in select symptomatic patients. At intravenous urography, the characteristic ‘paint brush’ appearance is due to the pooling of contrast, and linear striations radiating from the calyces.41,42 On ultrasound, the medullary pyramids appear uniformly hyperechoic due to the deposition of calcium in the affected tubules.1,43 On unenhanced CT, the presence of medullary nephrocalcinosis is commonly identified and CT urography demonstrates ‘papillary brush’ morphology and stones within the dilated collecting ducts (Figure 9).41,44 MSK has an excellent long term prognosis; however complications of recurrent urinary tract obstruction and infections may cause progressive renal failure.45
Multicystic dysplastic kidney
Multicystic dysplastic kidney (MCDK) is a non-hereditary developmental condition that results from urinary tract obstruction during embryogenesis with subsequent abnormal metanephric-mesenchymal differentiation.46,47 Flank pain, palpable mass and recurrent UTIs are common presenting symptoms. Imaging appearance of MCDK may vary depending on the age of the patients. Multiple, noncommunicating cysts in an enlarged, non-reniform shaped kidney without any functioning renal parenchyma is the typical appearance of MCDK in children.48 In adult patients, MCDK may appear as a multiloculated cystic mass mimicking a cystic neoplasm; however, a non-functioning kidney with peripherally located multiloculated cysts and absence of ipsilateral renal vessels, pelvicalyceal system, and ureter favor the diagnosis of MCDK (Figure 10).1 MCDK follows a benign course and the dysplastic kidney involutes over time.46,49
Localized renal cystic disease
Localized renal cystic disease (LRCD) is a rare, acquired, maldevelopmental condition characterized by multiple cysts replacing a variable portion of the kidney or the entire kidney with the cysts being separated by normal (or atrophic) renal tissue.50,51 Clinically, the affected individuals present with hematuria, hypertension, flank pain and palpable abdominal mass. At cross-sectional imaging, LRCD is characterized by conglomerate mass of multiple simple cysts involving one kidney, separated by normal / atrophic renal parenchyma (Figure 11).50,52 Differential diagnoses includes ADPKD, cystic renal tumor, and MCDK. While the presence of renal vessels and normal contrast excretion separates it from MCDK, unilaterality and the absence of extra-renal manifestations such as hepatic cysts differentiates it from ADPKD. Evaluation of entire kidney in coronal and sagittal planes helps in identification of normal renal parenchyma of LRCD from thickened septations of cystic renal neoplasms. 1 LRCD is a benign, nonsurgical condition that only requires imaging follow-up.50
Diagnosis of adult renal cystic disease: A practical approach
Renal size (large vs. normal vs. small), location and laterality of the cysts (cortex vs. medulla; unilateral vs. bilateral), and associated findings in other organs will help in making a definitive diagnosis in an adult patient with multiple renal cysts. Bilateral enlarged kidneys with multiple hemorrhagic and non-hemorrhagic cortical cysts with associated hepatic cysts are typical of ADPKD. Multiple, bilateral medullary cysts in small to normal sized kidneys in appropriate clinical setting favor the diagnosis of MCKD. Bilateral normal to mildly enlarged kidneys with complex renal cysts and multiple enhancing renal masses in conjunction with pancreatic cysts, central nervous system hemangioblastomas are characteristic of VHL. Multiple, bilateral renal angiomyolipomas intermixed with renal cysts in normal to enlarged kidneys is typically seen in Tuberous Sclerosis Complex patients. Bilateral small kidneys with multiple cysts in patients with end stage renal disease is diagnostic of ACKD. Medullary nephrocalcinosis with or without associated cystic dilatation of medullary collecting ducts in bilaterally small to normal sized kidneys is typically seen in MSK patients. Nonfunctioning unilateral kidney with multiloculated appearance and without identifiable renal vessels and pelvicalyceal system is characteristically seen in MCDK. Unilateral or segmental disease with conglomerate renal cysts intervening with normal or atrophic renal parenchyma that demonstrate normal contrast excretion is characteristic of LRCD. Among all, VHL and ACKD have significantly increased risk of renal cell carcinoma and require long-term surveillance.
Conclusion
There is a broad spectrum of hereditary and non-hereditary disorders that cause renal cystic disease in adults. Recent advances in the field of genetics and pathology have thrown fresh light on pathogenesis of some of these disorders leading to development of novel drugs aimed at halting disease progression. Cross-sectional imaging studies not only aid in the detection and differentiation of various chronic adult renal cystic diseases but also play a significant role in the surveillance and follow-up of patients. Improved awareness of adult renal cystic diseases and familiarity with their cross-sectional imaging findings permit appropriate diagnosis and patient management.
References
- Katabathina VS, Kota G, Dasyam AK, et al. Adult renal cystic disease:a genetic, biological, and developmental primer. Radiographics. 2010;30:1509-1523.
- Chapman AB, Wei W. Imaging approaches to patients with polycystic kidney disease. Semin Nephrol. 2011;31:237-244.
- Torra R, Darnell A, Cleries M, et al. Polycystic kidney disease patients on renal replacement therapy:data from the Catalan Renal Registry. Contrib Nephrol. 1995;115:177-181.
- Wilson PD, Goilav B. Cystic disease of the kidney. Annu Rev Pathol. 2007;2:341-368.
- Fliegauf M, Benzing T, Omran H. When cilia go bad:cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880-893.
- Zhang Q, Taulman PD, Yoder BK. Cystic kidney diseases:all roads lead to the cilium. Physiology (Bethesda). 2004;19:225-230.
- Ong AC, Harris PC. Molecular pathogenesis of ADPKD:the polycystin complex gets complex. Kidney Int. 2005;67:1234-1247.
- Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129-137.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008;359:1477-1485.
- Choyke PL. Inherited cystic diseases of the kidney. Radiol Clin North Am. 1996;34:925-946.
- Chang MY, Ong AC. Autosomal dominant polycystic kidney disease:recent advances in pathogenesis and treatment. Nephron Physiol. 2008;108:1-7.
- Chapman AB. Approaches to testing new treatments in autosomal dominant polycystic kidney disease:insights from the CRISP and HALT-PKD studies. Clin J Am Soc Nephrol. 2008;3:1197-1204.
- Bae KT, Grantham JJ. Imaging for the prognosis of autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2010;6:96-106.
- Truong LD, Choi YJ, Shen SS, et al. Renal cystic neoplasms and renal neoplasms associated with cystic renal diseases:pathogenetic and molecular links. Adv Anat Pathol. 2003;10:135-159.
- Bisceglia M, Galliani CA, Senger C, et al. Renal cystic diseases:a review. Adv Anat Pathol. 2006;13:26-56.
- Hildebrandt F, Otto E. Molecular genetics of nephronophthisis and medullary cystic kidney disease. J Am Soc Nephrol. 2000;11:1753-1761.
- Adams M, Smith UM, Logan CV, Johnson CA. Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet. 2008;45:257-267.
- Kiser RL, Wolf MT, Martin JL, et al. Medullary cystic kidney disease type 1 in a large Native-American kindred. Am J Kidney Dis. 2004;44:611-617.
- Auranen M, Ala-Mello S, Turunen JA, Jarvela I. Further evidence for linkage of autosomal-dominant medullary cystic kidney disease on chromosome 1q21. Kidney Int. 2001;60:1225-1232.
- Wolf MT, Mucha BE, Attanasio M, et al. Mutations of the Uromodulin gene in MCKD type 2 patients cluster in exon 4, which encodes three EGF-like domains. Kidney Int. 2003;64:1580-1587.
- Rizk D, Chapman AB. Cystic and inherited kidney diseases. Am J Kidney Dis. 2003;42:1305-1317.
- Scolari F, Viola BF, Prati E, et al. Medullary cystic kidney disease:past and present. Contrib Nephrol. 2001:68-78.
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059-2067.
- Hasani-Ranjbar S, Amoli MM, Ebrahim-Habibi A, et al. Mutation screening of VHL gene in a family with malignant bilateral pheochromocytoma:from isolated familial pheochromo cytoma to von Hippel-Lindau disease. Fam Cancer. 2009;8:465-471.
- Leung RS, Biswas SV, Duncan M, Rankin S. Imaging features of von Hippel-Lindau disease. Radiographics. 2008;28:65-79;quiz 323.
- Kuehn EW, Walz G, Benzing T. Von hippel-lindau:a tumor suppressor links microtubules to ciliogenesis and cancer development. Cancer Res. 2007;67:4537-4540.
- Wiesener MS, Maxwell PH, Eckardt KU. Novel insights into the role of the tumor suppressor von Hippel Lindau in cellular differentiation, ciliary biology, and cyst repression. J Mol Med. 2009;87:871-877.
- Chauveau D, Duvic C, Chretien Y, et al. Renal involvement in von Hippel-Lindau disease. Kidney Int. 1996;50:944-951.
- Meister M, Choyke P, Anderson C, Patel U. Radiological evaluation, management, and surveillance of renal masses in Von Hippel-Lindau disease. Clin Radiol. 2009;64:589-600.
- Narayanan V. Tuberous sclerosis complex: Genetics to pathogenesis. Pediatr Neurol. 2003;29:404-409.
- Rakowski SK, Winterkorn EB, Paul E, et al. Renal manifestations of tuberous sclerosis complex:Incidence, prognosis, and predictive factors. Kidney Int. 2006;70:1777-1782.
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345-1356.
- Bonnet CS, Aldred M, von Ruhland C, et al. Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum Mol Genet. 2009;18:2166-2176.
- Hartman TR, Liu D, Zilfou JT, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18:151-163.
- Umeoka S, Koyama T, Miki Y, et al. Pictorial review of tuberous sclerosis in various organs. Radiographics. 2008;28:e32.
- Choyke PL. Acquired cystic kidney disease. Eur Radiol. 2000;10:1716-1721.
- Levine E. Acquired cystic kidney disease. Radiol Clin North Am. 1996;34:947-964.
- Bhatnagar R, Alexiev BA. Renal-cell carcinomas in end-stage kidneys:a clinicopathological study with emphasis on clear-cell papillary renal-cell carcinoma and acquired cystic kidney disease-associated carcinoma. Int J Surg Pathol. 2012;20:19-28.
- Tickoo SK, dePeralta-Venturina MN, Harik LR, et al. Spectrum of epithelial neoplasms in end-stage renal disease:an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol. 2006;30:141-153.
- Gambaro G, Feltrin GP, Lupo A, et al. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease):a Padua Medical School discovery in the 1930s. Kidney Int. 2006;69:663-670.
- Maw AM, Megibow AJ, Grasso M, Goldfarb DS. Diagnosis of medullary sponge kidney by computed tomographic urography. Am J Kidney Dis. 2007;50:146-150.
- Prasad SR, Narra VR, Shah R, et al. Segmental disorders of the nephron:histopathological and imaging perspective. Br J Radiol. 2007;80:593-602.
- Levine E, Hartman DS, Meilstrup JW, et al. Current concepts and controversies in imaging of renal cystic diseases. Urol Clin North Am. 1997;24:523-543.
- Lang EK, Macchia RJ, Thomas R, et al. Multiphasic helical CT diagnosis of early medullary and papillary necrosis. J Endourol. 2004;18:49-56.
- Jungers P, Joly D, Barbey F, et al. ESRD caused by nephrolithiasis:prevalence, mechanisms, and prevention. Am J Kidney Dis. 2004; 44:799-805.
- Merrot T, Lumenta DB, Tercier S, et al. Multicystic dysplastic kidney with ipsilateral abnormalities of genitourinary tract:experience in children. Urology. 2006;67:603-607.
- Pope JC, Brock JW, 3rd, Adams MC, et al. How they begin and how they end:classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol. 1999;10:2018-2028.
- Jeon A, Cramer BC, Walsh E, Pushpanathan C. A spectrum of segmental multicystic renal dysplasia. Pediatr Radiol. 1999;29:309-315.
- Rabelo EA, Oliveira EA, Silva JM, et al. Conservative management of multicystic dysplastic kidney:clinical course and ultrasound outcome. J Pediatr. 2005;81:400-404.
- Slywotzky CM, Bosniak MA. Localized cystic disease of the kidney. AJR Am J Roentgenol. 2001;176:843-849.
- Hwang DY, Ahn C, Lee JG, et al. Unilateral renal cystic disease in adults. Nephrol Dial Transplant. 1999;14:1999-2003.
- Kim DJ, Kim MJ. Localized cystic disease of the kidney:CT findings. Abdom Imaging. 2003;28:588-592.
Citation
VS K, S V, K G, SR P.Update on adult renal cystic diseases. Appl Radiol. 2015; (11):44-50.
November 3, 2015