Multicystic Dysplastic Kidney
Images
Case Summary
A toddler had an ultrasound in early infancy showing a small right kidney. The child was also born with a vaginal cyst that regressed for a time and then recurred, prompting an imaging workup. The right kidney could no longer be identified on ultrasound.
Imaging Findings
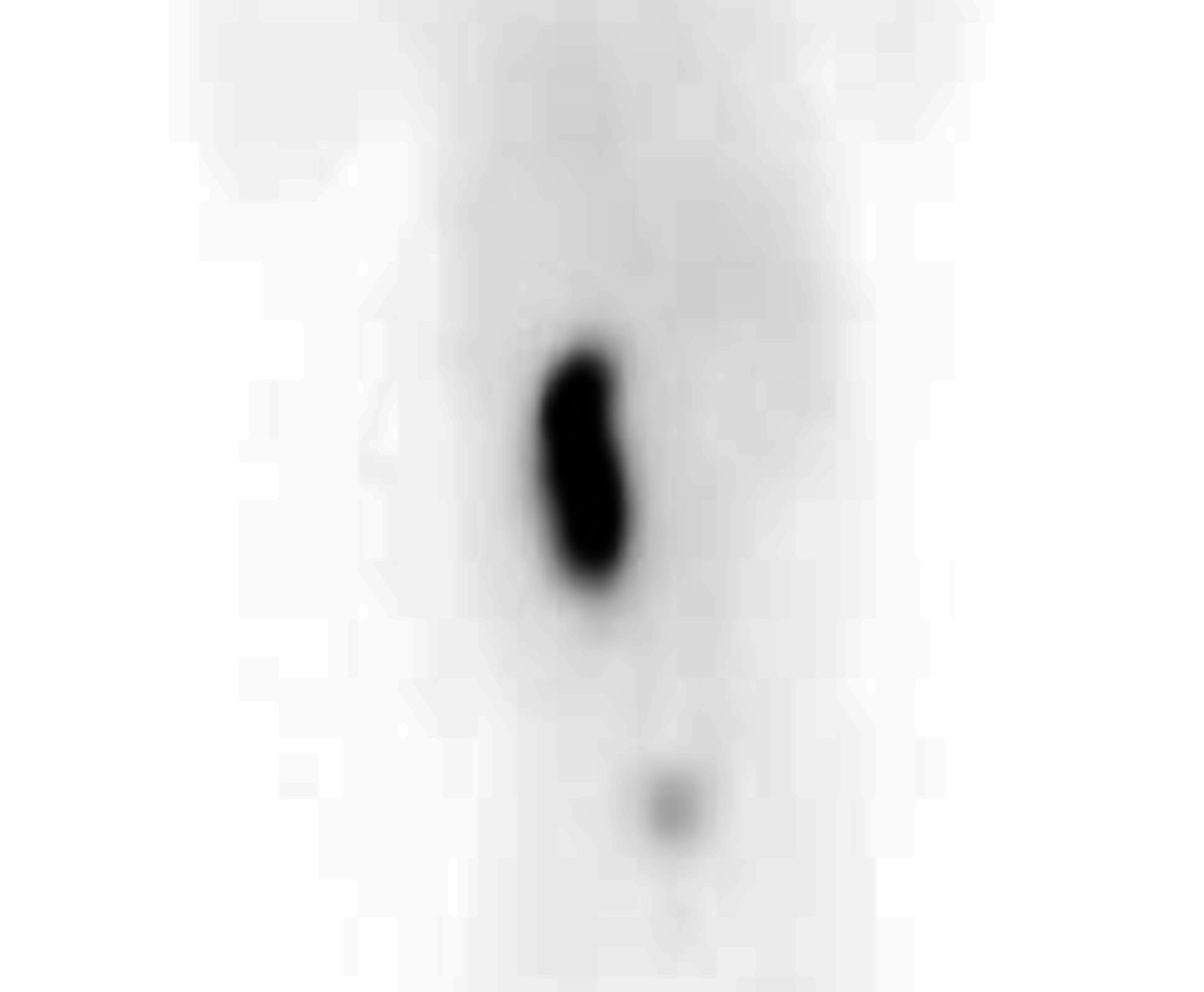
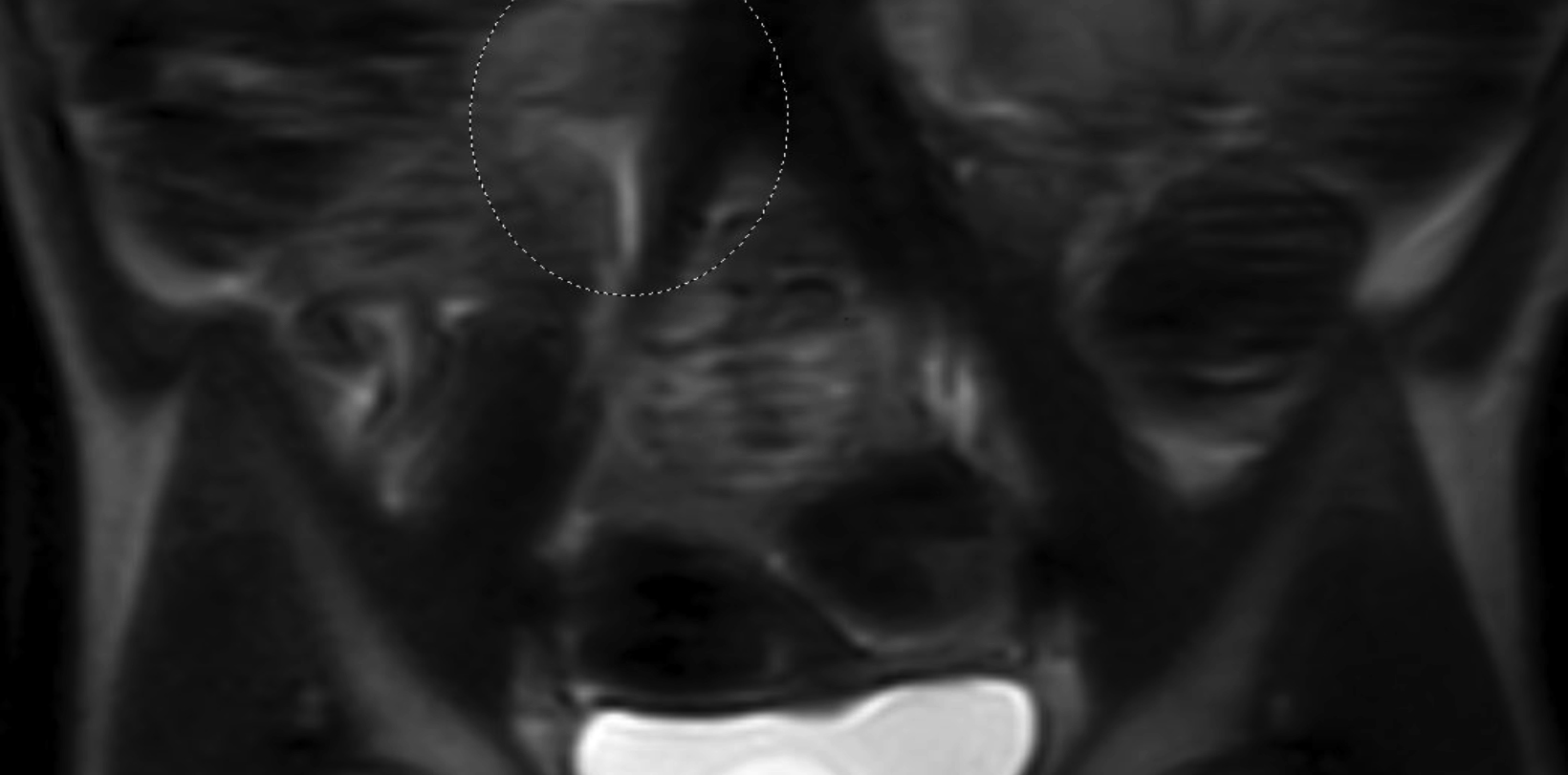
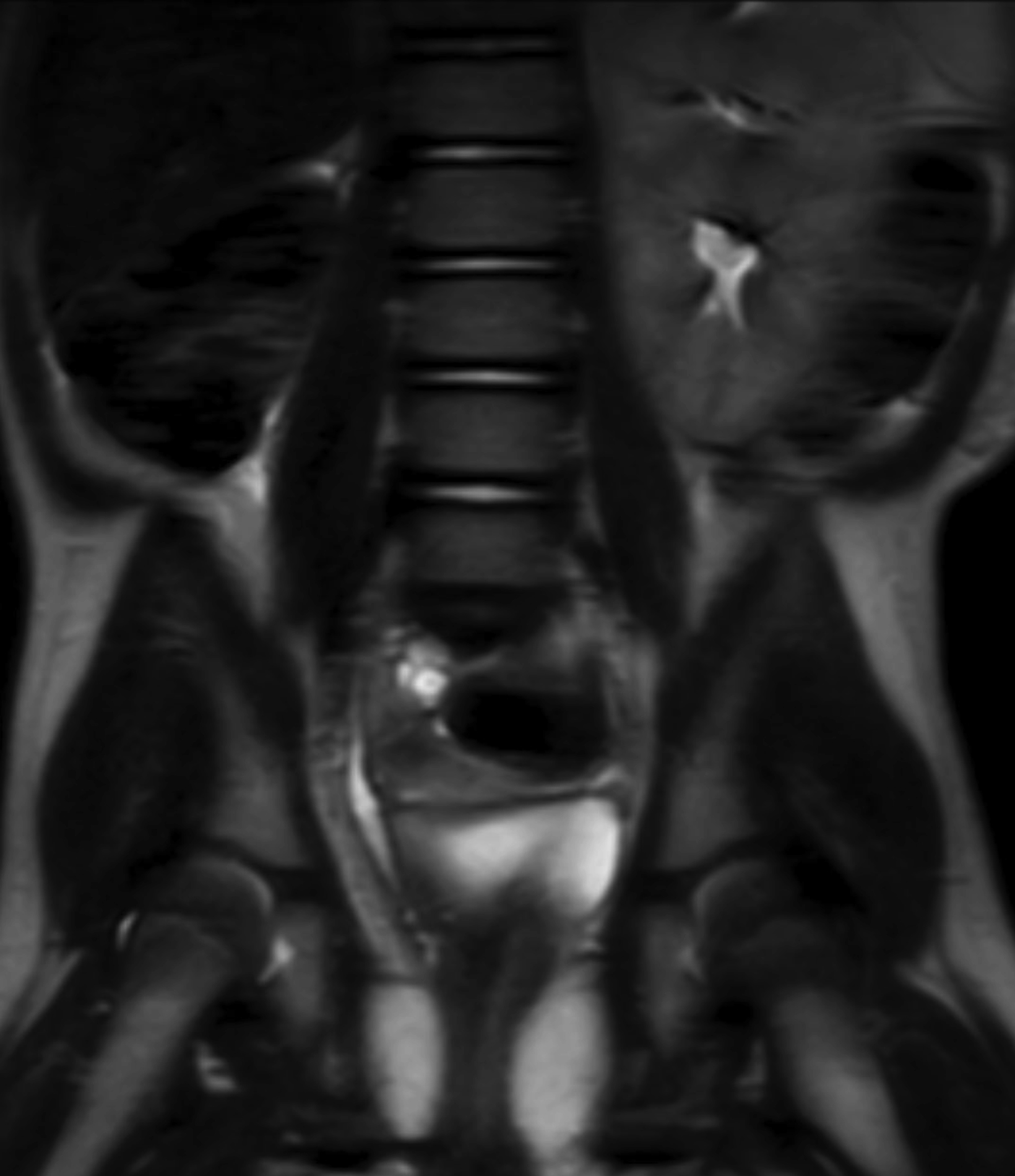
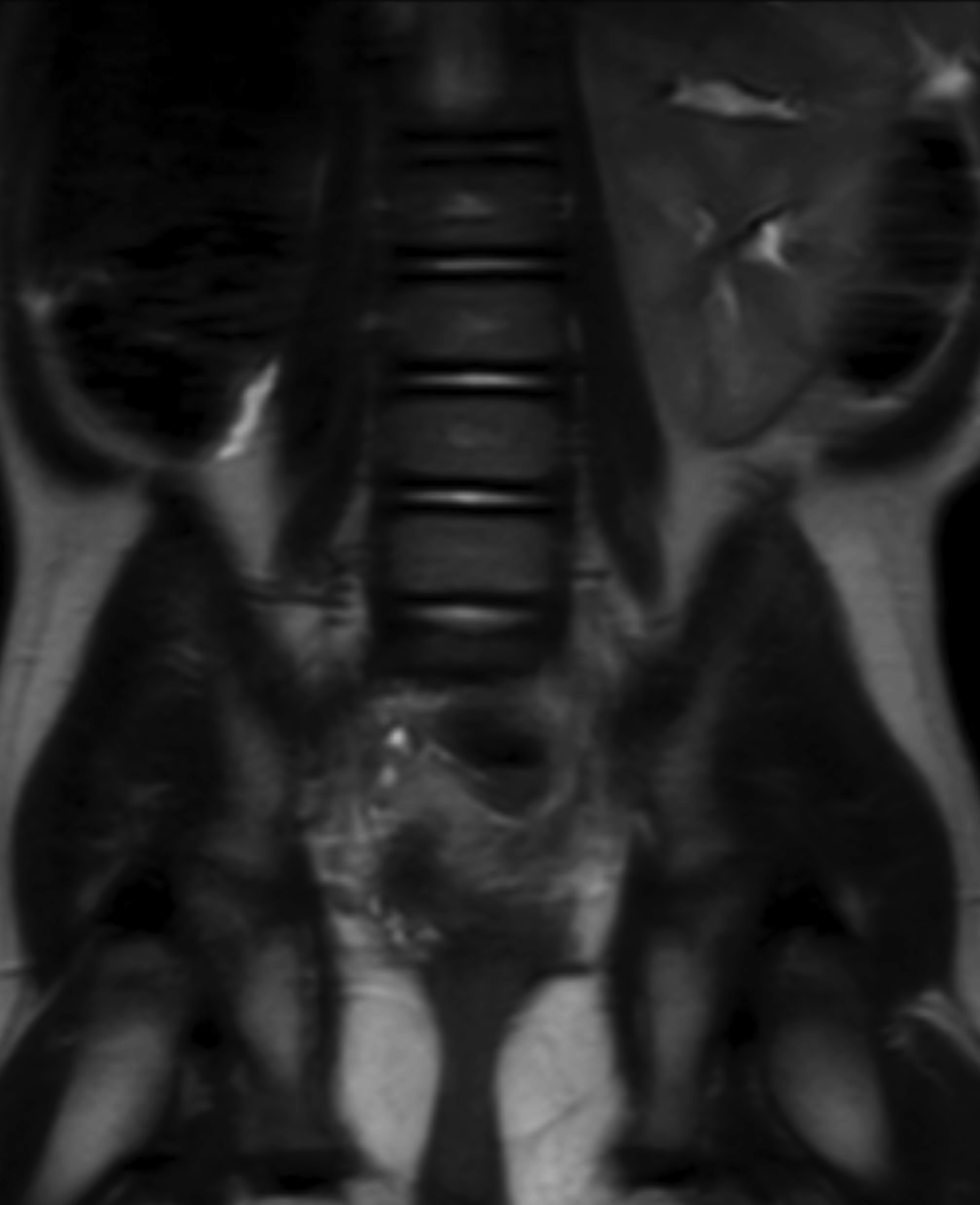
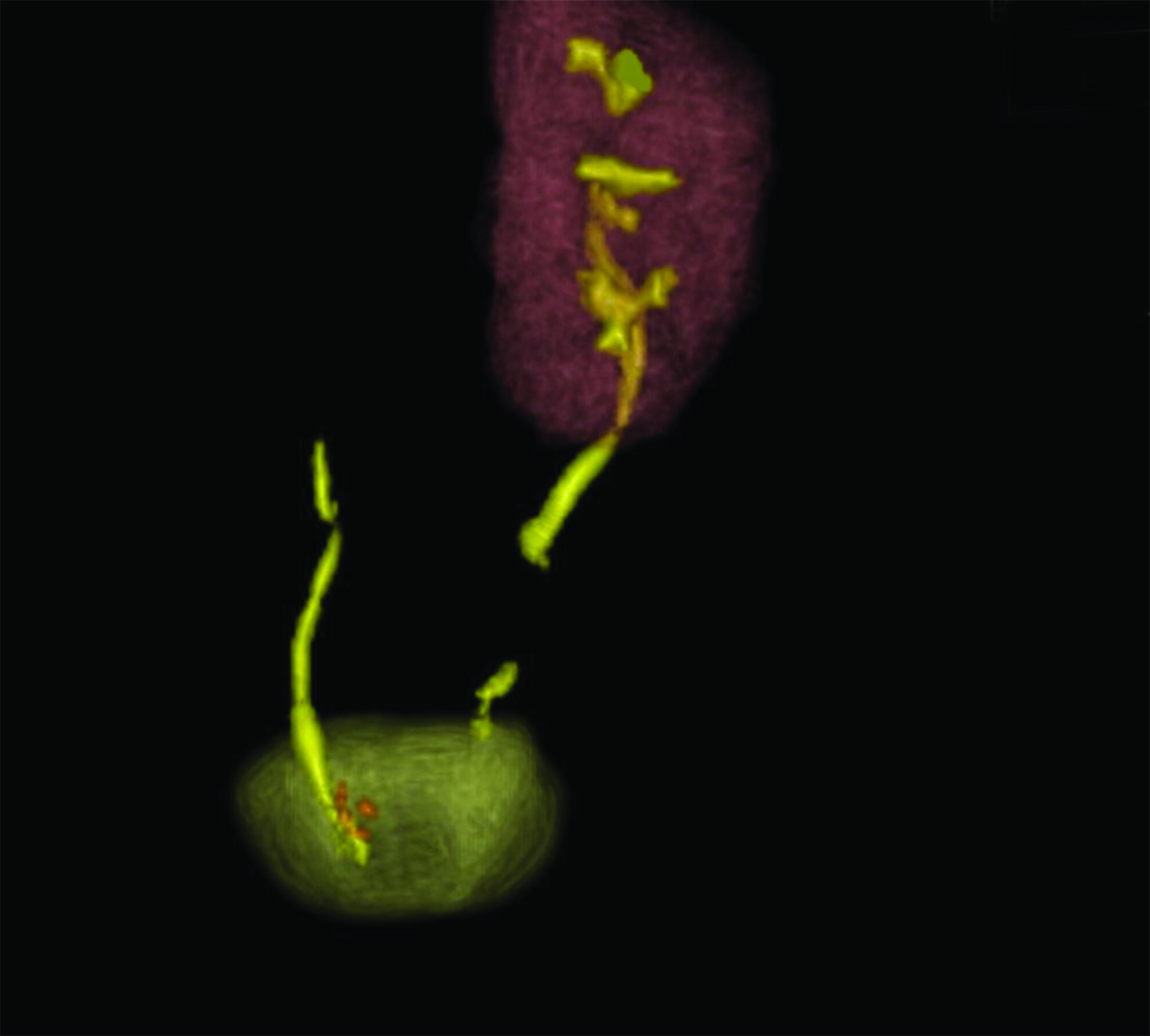
A posterior image from a Tc-99m MAG3 nuclear medicine renal study (Figure 1) showed a nonfunctioning right kidney and stasis of tracer-laden urine in the left renal collecting system. Coronal T2 magnetic resonance images (Figure 2) showed soft tissue of intermediate signal intensity, from the atrophic kidney, located anterior to the right psoas muscle, with a subjacent fluid filled tubular structure heading toward the lower pelvis. Small cystic structures were present on the right, anterolateral to the vagina and inferior to the bladder. The three-dimensional reconstructions from the MRI exam showed the right ureter, but not the right kidney (Figure 3).
Diagnosis
Multicystic dysplastic kidney. The differential diagnosis for non-visualized kidney includes renal agenesis, renal ectopia (pelvic kidney, crossed fused ectopia, horseshoe kidney1,2,3 ), and regression of a multicystic dysplastic kidney.
Discussion
Unilateral multicystic dysplastic kidney (MCDK) is a common abnormality, with an incidence of approximately 1 in 3640 births.4 The presence of an MCDK is often recognized on an antenatal ultrasound. Congenital cystic renal diseases overall occur in 2 to 4 in 1000 live births.5 Girls may present with incontinence from constant dribbling from an ectopic ureter insertion into the vagina. This may only be apparent during or after toilet training.
The pathogenesis of MCDK is incompletely understood but likely involves a complex interaction of multiple features, including gene expression, ciliary abnormalities, and embryologic development. Multicystic dysplastic kidneys (are associated with ureteropelvic junction obstruction and vesicoureteral reflux in the contralateral kidney in a variable percentage of children.4,5 They can be associated with ectopic ureteral insertion, as in this case.
Abdominal ultrasound performed early in life shows multiple noncommunicating renal cysts without a dilated renal pelvis in combination with abnormal or absent renal parenchyma. The renal parenchyma may be echogenic, have thinned cortex, and demonstrate poor corticomedullary differentiation. With time, the affected kidney becomes smaller and less visible before finally involuting. It is also important to image the contralateral kidney, as it will be the sole provider of renal function. Typically, this kidney shows compensatory hypertrophy. While the affected kidney typically does not function, some components may continue to produce urine. In questionable cases a technetium-99m MAG 3 renal scan can be used to assess renal function.
MRI and/or MR urography can be used to better evaluate complex urinary anomalies. While usually unnecessary in the setting of MCDK, MRI can be used in the setting of a patient who is constantly dribbling urine or in whom pelvic cysts are thought to be related to a renal anomaly.6 MCDK may be associated with other congenital anomalies or diagnosed in a child with a family history of renal abnormality (such as a solitary kidney). These patients are usually managed conservatively, with imaging follow-up dictated by the clinical presentation.7 Imaging protocols and timing are currently not standardized.
Conclusion
In a female infant or toddler presenting with a pelvic cyst associated with the urinary tract or vagina, or in an older girl present- ing with incontinence, an ectopic ureteral insertion should be considered. While an ectopic ureter insertion most often arises from a duplicated upper pole moiety, an ectopic ureter arising from an MCDK may be another consideration, as presented in this case.
References
- Avni FE, Garel C, Cassart M, et al. Imaging and classification of congenital cystic renal diseases. Am J Roentgenol. 2012;198: 1004-1013. doi: 10.2214/AJR.11.8083.
- Moyle PL, Kataoka MY, Nakai A, Takahata A, Reinhold C, Sala E. Nonovarian cystic lesions of the pelvis. Radiographics. 2010;30(4):921-938. doi:10.1148/rg.304095706.
- Aslam M and Watson AR. Unilateral multicystic dysplastic kidney: long term outcomes. Arch Dis Child. 2006: 91(10):820-823. doi: 10.1136/adc.2006.095786.
- Coley BD, editor-in-chief. Section 7: Genitourinary system. In: Caffey’s Pediatric Diagnostic Imaging, 13th edition. Philadelphia, PA: Elsevier. 2019: 1065-1218.
- Blumer SL, Biko DM, Halabi SS. Pediatric Genitourinary Tract. In: Pediatric Imaging: A Core Review. Philadelphia, Baltimore, New York, London, Buenos Aires, Hong Kong, Sydney, Tokyo. Wolters Kluwer; 2019. 45-82.
- Towbin R, Martin L. Multilocular cystic dysplasia of half of a horseshoe kidney. J Pediatr Surg. 1974;9(3):421. doi:10.1016/s0022-3468(74)80308-8.
- Alam K, Varshney M, Aziz M, et al. Multicystic Renal Dysplasia. Brit Med J Case Rep. 2011. doi: 10.1136/bcr.03.2011.3989.
Citation
S H, SR L, SA J, AJ T, RB T.Multicystic Dysplastic Kidney. Supplement to Applied Radiology. 2022; (6):6-8.
November 2, 2022