Malignant Ectomesenchymoma of the Scalp
Images

CASE SUMMARY
A 2-year-old with a history of hypoxic-ischemic injury and chronic hydrocephalus presented with a rapidly growing and highly vascularized scalp mass. Following preoperative embolization, the tumor was completely resected. Pathologic analysis revealed a malignant ectomesenchymoma (MEM), a rare tumor that occurs most frequently in children.
IMAGING FINDINGS
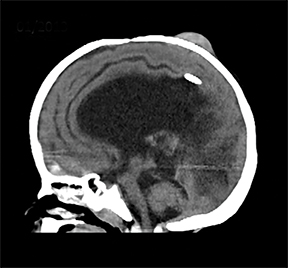
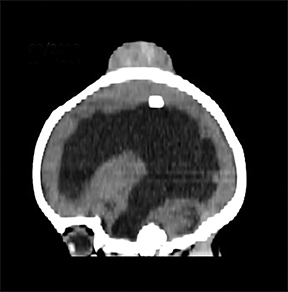
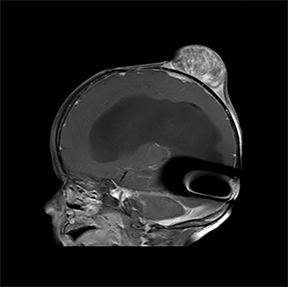
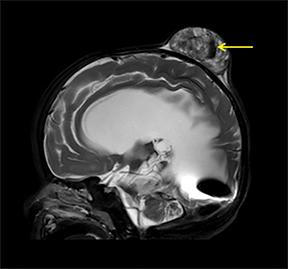
A computed tomography (CT) scan of the head without contrast (Figure 1) showed a solid, heterogeneous mass in the deep subcutaneous tissues of the scalp near the vertex. The mass showed significant growth over a 4-week period. The underlying bone was thin and irregular without frank cortical disruption. On magnetic resonance imaging (MRI, Figure 2), the mass was isointense to hypointense on T1 (Figure 2A), heterogeneous on T2 (Figure 2B) and showed patchy contrast enhancement (Figure 2C). On MR angiography, prominent branches from the right superficial temporal artery extended to the lesion core (Not shown).
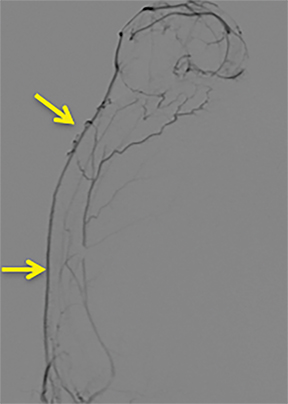
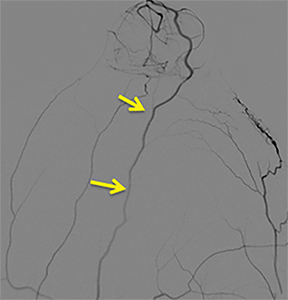
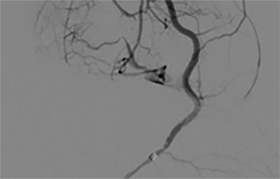
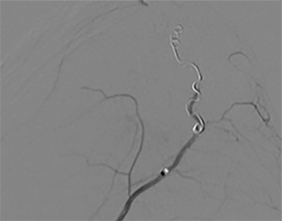
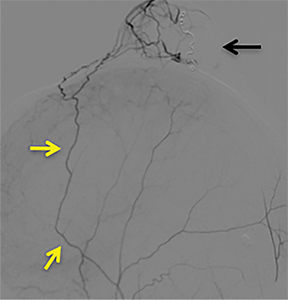
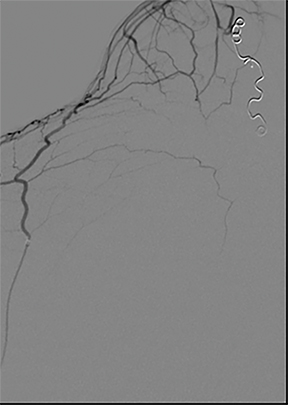
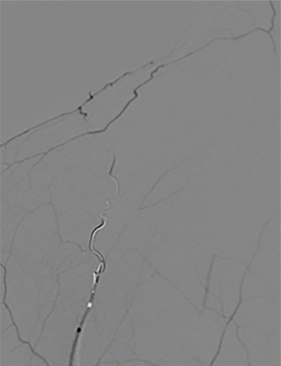
A cerebral angiogram was subsequently performed for preoperative embolization. The frontal and lateral pre- and postembolization images showed the main arterial supply and near-complete occlusion after coil embolization (Figures 4, 5).
DIAGNOSIS
Malignant ectomesenchymoma of the scalp. Differential diagnosis includes rhabdomyosarcoma, neuroblastoma, lymphoma, neuroepithelioma, extraskeletal Ewing sarcoma, and other round-cell sarcomas.
DISCUSSION
Malignant ectomesenchymomas are rare neoplasms believed to arise from widely distributed, pluripotential, migratory neural crest cells that form neuroectodermal and mesenchymal elements. These lesions usually affect young children and show rapid interval growth.
On microscopic examination, the predominant tumor component is mesenchymal. Rhabdomyosarcoma is the most common mesenchymal element in children, while other mesenchymal elements can dominate in adults. The neural elements can be scattered or form clusters of ganglion or more primitive neuroblastic/neuroectodermal cells.
The soft tissues of the head and neck, scrotum, extremities, and pelvic, and retroperitoneal regions are common sites of involvement for MEM. To date and to our knowledge, only a few intracranial tumors have been reported.
Imaging findings for these tumors are not specific; the differential diagnosis is based on patient age and tumor location. Most neoplasms are well circumscribed: gray, white, or tan and composed of firm tissue with a variable degree of necrosis. The available histological data define ganglioneuroma as the most common neural element, followed by ganglion cells, neuroblastomas, and other neural elements. The optimal treatment has not been established due to its rarity and lack of supporting data. Published cases support surgery as the mainstay of therapy based on the fact that most MEM survivors have had a complete tumor resection at diagnosis.
The roles of chemotherapy and radiation have not been well established. However, most pediatric MEM cases are treated using a multimodality approach. The choice of chemotherapy regimen is based on the rationale that treatment of biphenotypic tumors should be focused on the most aggressive component. Most cases of extracranial MEM have been treated according to the Intergroup Rhabdomyosarcoma Study Group trials.
The prognosis of MEM depends on tumor site, size, resectability, dissemination, and achievement of remission. The best therapeutic results are seen with complete tumor resection and pre- or postsurgical chemotherapy. Owing to our patient’s overall poor prognosis, further treatment was not pursued,
CONCLUSION
Malignant ectomesenchymomas, part of the heterogeneous group of soft tissue sarcomas, are rare tumors in the pediatric population. They are composed of neural and mesenchymal elements and may present as intra- or extracranial neoplasms.
The appropriate histological, immunohistochemical, and ultra-structural analysis are critical to ruling out other diagnostic possibilities, including rhabdomyosarcoma, neuroblastoma, lymphoma, neuroepithelioma, extraskeletal Ewing sarcoma and other round-cell sarcomas.
The recommended therapy is complete resection followed by chemotherapy directed against the most aggressive tumor component which, in the majority of pediatric patients, is rhabdomyosarcoma. The prognosis of these tumors is comparable to other rhabdomyosarcoma-like soft-tissue tumors if treated in similar fashion.
REFERENCES
- Miller HL, Marx A, Trusen M, et al. Disseminated Malignant Ectomesenchymoma (MEM): Case Report and review of the literature. Pediatr Hematol Oncol (2002); 19:9-17.
- Dantonello TM, Leuschner I, Vokuhl C, et al. Malignant Ectomesenchymoma in Children and Adolescents: Report from the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer (2013);60:224-229.
- Altenburger DL, Wagner AS, Eslin DE, et al. A rare case of malignant ectomesenchymoma arising from the falx cerebri. Case Report. J Neurosurg Pediatrics (2011); 7:94-97.
- Weiss E, Albrecht CF, Herms J, et al. Malignant ectomesenchymoma of the cerebrum. Case Report and discussion of therapeutic options. Eur J Pediatr (2005); 164:345-349.
- Freitas A, Aguiar P, Miura F, et al. Malignant Ectomesenchymoma. Case Report and Review of the Literature. Pediatr Neurosurg (1999); 30:320-330
Citation
P C, J E, R K, R B, D D, R A, RB T.Malignant Ectomesenchymoma of the Scalp. Appl Radiol. 2020; (3):40-42.
May 5, 2020