Giant axonal neuropathy
Images
![]()
CASE SUMMARY
A 9-year-old white male presented with progressive abnormal gait and tightly curled scalp hair. Magnetic resonance imaging revealed a leukodystrophy pattern involving the cerebellar and supratentorial white matter.
IMAGING FINDINGS
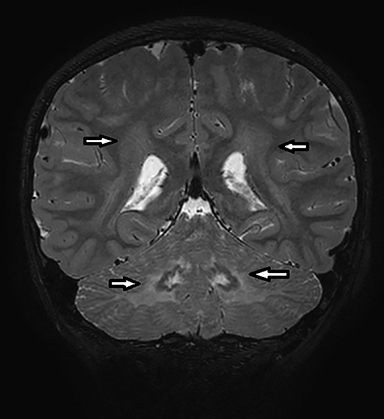
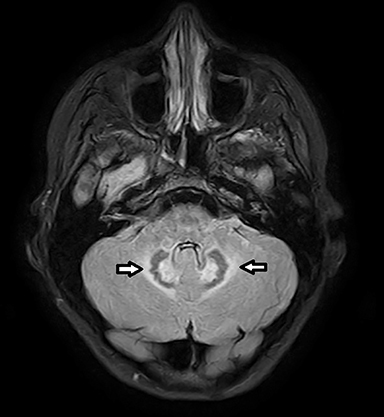
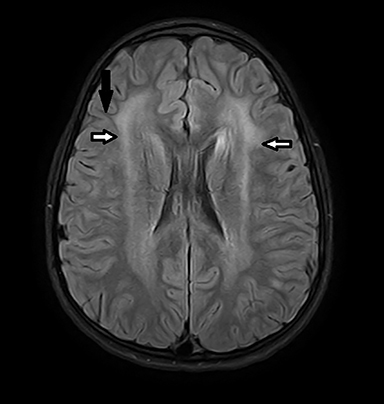
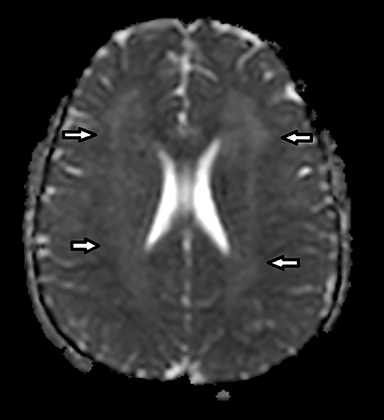
Coronal T2-weighted image (Figure 1) shows symmetric alternating increased signal in the hila, normal hypointensity of the dentate nuclei and surrounding hyperintensity of the cerebellar white matter. Axial FLAIR image (Figure 2) through the cerebellum accentuates the T2 findings. Axial FLAIR image (Figure 3) through the centrum semiovale shows confluent white matter hyperintensity with sparing of subcortical U fibers. Axial ADC map (Figure 4) shows diffusely increased diffusivity within abnormal white matter. No evidence of restricted diffusion.
DIAGNOSIS
Imaging findings are that of a nonspecific leukodystrophy with sparing of subcortical U fibers and early cerebellar involvement. The diagnosis of giant axonal neuropathy (GAN) was confirmed by clinical examination and genetic testing.
Krabbe disease may mimic GAN with early cerebellar involvement and is more common. Metachromatic leukodystrophy is more common than both Krabbe and GAN, but does not commonly show cerebellar involvement until late in the disease. Both Krabbe and Metachromatic leukodystrophy spare subcortical U fibers.
DISCUSSION
Giant axonal neuropathy (GAN) is a rare, autosomal recessive, early onset, fatal neurodegenerative disorder that affects both the peripheral and central nervous system.1,2 The disease stems from gigaxonin gene mutations resulting in disorganization of axonal intermediate filaments.2 Pronounced peripheral motor and sensory neuropathy begins in infancy, and the disease progresses centrally with cognitive impairment, seizures and pyramidal tract signs.1 Patients become wheelchair bound in the second decade and eventually bedridden with severe polyneuropathy, ataxia and dementia. Death usually occurs in the third decade.1 The disease has no sex predilection. On clinical examination there is tightly curled scalp hair that is profoundly different from the parents’ hair.1
CT may show supratentorial or cerebellar white matter hypodensity, however CT is often normal. MRI is the modality of choice when clinical suspicion is high, and most commonly shows a nonspecific leukodystrophy.2,3 Confluent white matter T2/FLAIR hyperintensity involving frontal and parietal white matter with sparing of subcortical U fibers is the hallmark of GAN.3 T2 hyperintensity is also seen in the pons, middle cerebellar peduncles, and bilateral cerebellar white matter. 3 Posteromedial thalamus may also be affected.3 The globus pallidus may demonstrate hyperintensity on both T1- and T2-weighted images.3,4 Increased apparent diffusion coefficient values are seen in abnormal white matter, basal ganglia and thalamus secondary to loss of myelin.5 However, the corpus callosum is spared.3 Parenchymal volume loss including cervical cord volume loss may be seen.3 Spectroscopy demonstrates elevation of choline peaks and lactate indicative of demyelination.3 Reduction of NAA is indicative of neuronal loss. Spectroscopy ratios show that there is more severe cerebellar involvement, which corresponds with pathological data.3
The diagnosis of GAN is suggested by clinical findings, nerve conduction studies and MRI, and is established by immunodiagnostic testing. Sural nerve biopsy is no longer routinely used.1 Traditional treatment focuses on optimizing intellectual and physical development through a multidisciplinary approach with involvement of neurologists, orthopedic surgeons, psychologists, speech and occupational therapists. Physiotherapy is utilized to preserve mobility as long as possible.1 Gene therapy trials were initiated in February of 2015 and are currently in Phase I trials slated to be completed in July 2018 as of this publication (https://clinicaltrials.gov/ct2/show/NCT02362438).
CONCLUSION
Giant axonal neuropathy is a rare, fatal neurodegenerative disorder that often manifests on imaging as a nonspecific leukodystrophy. However, GAN should be considered among a short list of diagnostic considerations in the setting of early cerebellar involvement and sparing of subcortical U fibers. GAN may mimic Krabbe disease; however, it commonly carries a dramatically different clinical picture. With sural nerve biopsy becoming less common, there is a growing dependence on genetic testing to establish the diagnosis.
REFERENCES
- Kuhlenbaumer G, Timmerman V, Bomont P. Giant axonal neuropathy. Gene Reviews. [internet]. https://www.ncbi.nlm.nih.gov/books/NBK1136/ Published January 9, 2003. Updated October 9, 2014. Accessed November 2016.
- Israni A, Chakrabarty B, Gulati S, et al. Giant axonal neuropathy: A clinicoradiopathologic diagnosis. Neurology.2014; 82: 816-817.
- Ravishankar S, Goel G, Rautenstrauss C, Nalini A. Spectrum of magnetic resonance imaging findings in a family with giant axonal neuropathy confirmed by genetic studies. Neurology India. 2009; 57(2): 181-184.
- Erol I, Alehan F, Alkan O, Bruno C. Involvement of the globus pallidus in giant axonal neuropathy. Pediatric Neurology. 2012; 47(5): 382-384.
- Alkan A, Sigirci A, Kutlu R, Doganay S, Erdem G. Giant axonal neuropathy: diffusion-weighted imaging features of the brain. J Child Neurol. 2006; 21(10): 912-915.
Citation
R C, SA J, AJ T, R T.Giant axonal neuropathy. Appl Radiol. 2018; (5):40-41.
May 8, 2018