Congenital Adrenal Hyperplasia
Images

CASE SUMMARY
An infant with ambiguous genitalia was born at 38 6/7 weeks’ gestation to consanguineous parents. Serum 17-OH progesterone obtained on the day of birth showed a value of 8,590 ng/dL (reference range: 1,000-3,000 ng/dL), followed by a value of 6, 240 ng/dL three days later. Subsequent genetic testing showed a 46, XX karyotype.
Physical examination revealed a “pseudoscrotum” without clefts, indicating the presence of fused labia. In addition, no testes were identified either in the pseudoscrotum or inguinal canal. A male-appearing phallus with chordee secondary to a midline ventral raphe with hypospadias was present. This was later confirmed to represent cliteromegaly.
There were signs of hyper-androgenization, including significant facial hair and cliteromegaly with a large clitoral base-to-anus ratio. Additionally, there was hyperpigmentation of the phallus suggestive of adrenocorticotropic hormone (ACTH) excess.
IMAGING FINDINGS
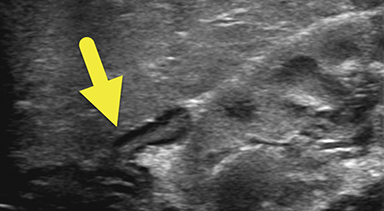
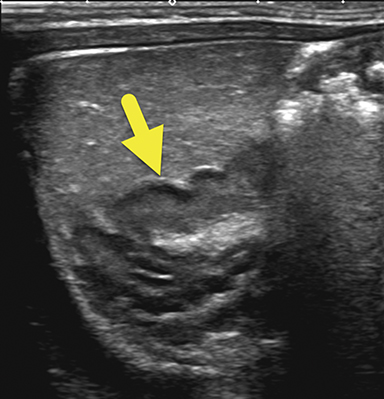
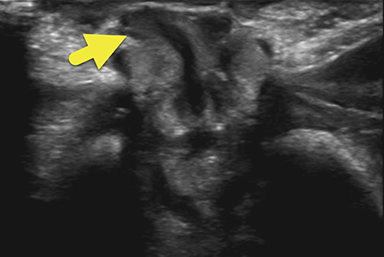
An ultrasound scan (US) at 12 hours of age (Figure 1) showed diffuse nonmass-like enlargement of the adrenal glands. While the adrenal glands maintained their adreniform shape, the body and arms of the glands had multiple folds, giving the glands a cerebri form appearance. Images of the pelvis and pseudoscrotum showed no identifiable testes, uterus, or ovaries. At the perineum a band-like hypoechoic structure was seen to extend into the phallus.
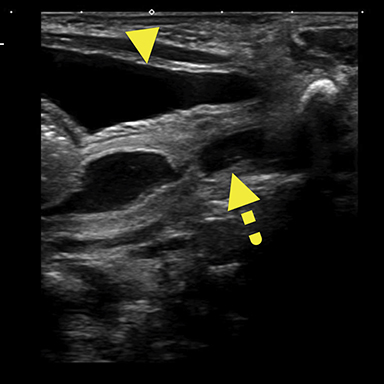
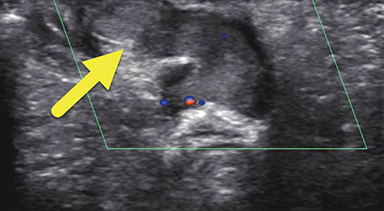
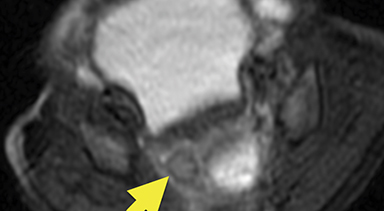
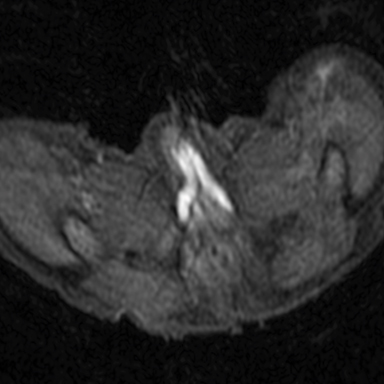
Magnetic resonance imaging (MRI) was performed in an attempt to identify gonadal structures (Figure 2). Again, no identifiable testes, uterus, or ovaries were present. An oval-shaped structure was present in the deep pelvis adjacent to the right side of the rectum. The phallus was composed of three cylindrical columns of tissue.
DIAGNOSIS
Congenital adrenal hyperplasia (CAH) resulting from a 17α hydroxylase deficiency
DISCUSSION
Congenital adrenal hyperplasia (CAH) is the most common cause of ambiguous genitalia. It is an enzyme deficiency disorder that manifests as defective steroidogenesis within the adrenal cortex.1 Enzyme deficiency disorders such as CAH are often the result of autosomal recessive inheritance patterns, for which consanguineous marriages are at an increased risk.2 There are five enzyme-mediated steps to synthesize cortisol from cholesterol in the zona fasciculata layer within the adrenal cortex.3 A mutation in any one of the enzymes that catalyzes these reactions can lead to CAH. The most common enzyme deficiency, found in 95% of patients with CAH, is 21-hydroxylase (21-OH),3 followed by 11 β-hydroxylase and 17α hydroxylase deficiency.
The enzyme 21-OH is responsible for the conversion of 17-hydroxyprogesterone (17-OHP) into 11-deoxycortisol, which is further converted into cortisol.3 The 21-OH enzyme also catalyzes the reaction conversion of progesterone into 11-deoxy corticosterone, a precursor to aldosterone. Clinically, CAH resulting from a 21-OH deficiency can be identified by decreased aldosterone and cortisol, and increased androgens.
This explains the clinical indication for obtaining the concentration of 17-OHP. The 17-OHP test reveals a 21-OH deficiency because it will reveal a high concentration of 17-OHP. Impaired cortisol synthesis leads to increased ACTH secretion from the anterior pituitary gland due to loss of negative feedback mechanisms regulated by cortisol.1, 4 The excess ACTH levels chronically activate the adrenal cortex, resulting in hyperplasia, increased production of cortisol precursor molecules, and increased synthesis of adrenal androgens.1 The elevated secretion of androgens results in clinical manifestations such as genital ambiguity, early onset of puberty, hirsutism, or oligomenorrhea.5
There are two types of 21-OH CAH based on symptom severity: non-classic and classic. Non-classic CAH is more common and less severe than classic CAH. Non-classical CAH patients may be either asymptomatic or present with less severe symptoms than classical CAH, such as ambiguous genitalia, hirsutism, menstrual irregularities, or other symptoms of androgen excess.6 The symptoms of non-classical CAH also may not develop until adolescence or adulthood.6
In the classic form of CAH, females present at birth with virilization of external genitalia owing to adrenal androgen exposure during sexual differentiation of the fetus; however, they have normal internal genitalia.1 Classical CAH patients are likely to experience early onset of puberty, hirsutism, and oligomenorrhea.5 The feared complication of salt-wasting is seen in 75% of patients with classic CAH.1 Salt wasting results from impaired synthesis of the salt-retaining steroid, aldosterone. Severely low aldosterone levels lead to decreased sodium reabsorption and decreased potassium secretion at the distal tubules of the kidney. This leads to hypotension, hyponatremia, hyperkalemia, vomiting, dehydration, and failure to thrive. In infants, salt wasting can cause shock, vascular collapse, and even death if not carefully managed.
The enzyme 11 β-hydroxylase is responsible for converting 11-deoxy cortisol into cortisol. It also catalyzes the reaction converting 11-deoxy corticosterone into corticosterone, a precursor to aldosterone. CAH caused by an 11 β-hydroxylase deficiency can be identified by decreased aldosterone and cortisol, as well as increased 11-deoxy corticosterone and androgens. Patients with an 11 β-hydroxylase deficiency present with the same symptoms as 21-OH CAH patients and are similarly categorized as either classical or non-classical. Patients with the classic form of 11 β-hydroxylase deficiency also present with hypertension caused by the increased 11-deoxy corticosterone.
The enzyme 17α hydroxylase converts pregnenolone into 17α-hydroxy pregnenolone, a precursor to cortisol. It also catalyzes the reaction converting progesterone into 17α-hydroxy progesterone, another precursor to cortisol. Congenital adrenal hyperplasia resulting from a 17α hydroxylase deficiency can be identified by elevated aldosterone, 11-deoxy corticosterone, and corticosterone, as well as decreased cortisol, androgens, and 17- hydroxylated precursors.8 Although the aldosterone pathway is elevated, aldosterone production is lower because of the elevated blood pressure caused by the higher levels of 11-deoxy corticosterone and corticosterone. Patients will experience hypokalemia and metabolic alkalosis owing to the increased mineralocorticoids.
Imaging is used to confirm a diagnosis of CAH, with US typically considered the first-line modality. Normally, the neonatal adrenal gland is enlarged, with a smooth contour and a thick hypoechoic adrenal cortex. The larger size and greater cortical thickening of the normal neonatal adrenal gland is related to cortisol production required for birth. Patients with CAH have an even larger adrenal gland than normal. The adrenal gland becomes so large that it begins to fold back upon itself. This back-folding gives the adrenal gland an irregular contour and its characteristic cerebriform shape.
Cross-sectional imaging via CT or MRI is generally not required. In this case, MRI was performed to identify sex organs, thus providing the parents with relevant information as they began caring for their child.
Glucocorticoid and mineralocorticoid replacement therapy is the main treatment of CAH. Our patient specifically was given replacement NaCl therapy, hydrocortisone, and fludrocortisone to prevent salt wasting, a potentially fatal complication of CAH.
With optimal treatment and prevention of comorbidities, CAH patients can achieve a normal life expectancy and quality of life, including normal linear growth and puberty development, regularization of menses, prevention of hirsutism, and increased fertility.6 Early diagnosis and treatment provide the best outlook for the patient; CAH can be diagnosed prenatally via amniocentesis or chorionic villus sampling, and can be effectively treated in utero.9
In a study of 532 prenatally diagnosed cases of CAH, New et al found that prenatal treatment with dexamethasone significantly reduced or eliminated virilization in females with classic 21-OH CAH.9 Long-term complications of CAH relate to fertility challenges, reduced bone mineral density, and increased cardiovascular risk and arrhythmias, neuropsychological issues, and height.10
CONCLUSION
Congenital adrenal hyperplasia is an autosomal recessive enzyme deficiency disorder. The disease process is specific to which enzyme in the corticosteroid pathway is mutated. The degree of severity is based on the level of available enzyme. CAH can be diagnosed prenatally via amniocentesis or chorionic villus sampling, or after birth using serum blood tests and ultrasonography. Patients with CAH are closely monitored during glucocorticoid and mineralocorticoid replacement therapy to prevent salt wasting.
REFERENCES
- New, M., et al., Congenital Adrenal Hyperplasia, in Endotext, K.R. Feingold, et al., Editors. 2000: South Dartmouth (MA).
- Sheikh Alshabab, L.I., et al., Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: A five-year retrospective study in the Children’s Hospital of Damascus, Syria. Qatar Med J, 2015. 2015(1): p. 11.
- Dessinioti, C. and A. Katsambas, Congenital adrenal hyperplasia. Dermatoendocrinol, 2009. 1(2): p. 87-91.
- Dutt, M. and I. Jialal, Physiology, Adrenal Gland, in StatPearls. 2019: Treasure Island (FL).
- Pignatelli, D., S.S. Pereira, and R. Pasquali, Androgens in Congenital Adrenal Hyperplasia. Front Horm Res, 2019. 53: p. 65-76.
- Witchel, S.F. and R. Azziz, Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol, 2010. 2010: p. 625105.
- Tusie-Luna, M.T., P. Traktman, and P.C. White, Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem, 1990. 265(34): p. 20916-22.
- Peter, M., W.G. Sippell, and H. Wernze, Diagnosis and treatment of 17-hydroxylase deficiency. J Steroid Biochem Mol Biol, 1993. 45(1-3): p. 107-16.
- New, M.I., et al., Prenatal diagnosis for congenital adrenal hyperplasia in 532 pregnancies. J Clin Endocrinol Metab, 2001. 86(12): p. 5651-7.
- Kamoun, M., et al., Congenital adrenal hyperplasia: Treatment and outcomes. Indian J Endocrinol Metab, 2013. 17(Suppl 1): p. S14-7.
Citation
TH R, RB T, AJ T.Congenital Adrenal Hyperplasia . Appl Radiol. 2021; (1):56C-56F.
January 19, 2021