Klippel-Feil Syndrome Essentials, Part 1: Embryological Development and Genetic Mechanisms
Editor’s note: This is Part 1 of a 2-part series. Part 2 will be featured in the January/February 2025 edition of Applied Radiology .
Introduction
Klippel-Feil syndrome (KFS) is a rare congenital fusion of the cervical spinal column, sometimes with concurrent involvement of the thoracic and lumbar vertebrae ( Figure 1 ).1 - 5 It was first described by Maurice Klippel and Andre Feil in 1912, with the presence of 3 classical clinical signs. The triad, which is seen in some but not all patients, consists of a short neck, limited neck movement, and low posterior hairline.2
Vertebral anomalies and multilevel rib fusion. The chest radiograph demonstrates multilevel lower cervical and upper thoracic vertebral anomalies (black arrows) and multilevel rib fusion (white arrows) seen in Klippel-Feil syndrome (KFS). It also depicts right-sided elevation of the scapula and an omovertebral bone (inferior to black arrow), correlated with both KFS and Sprengel deformity.
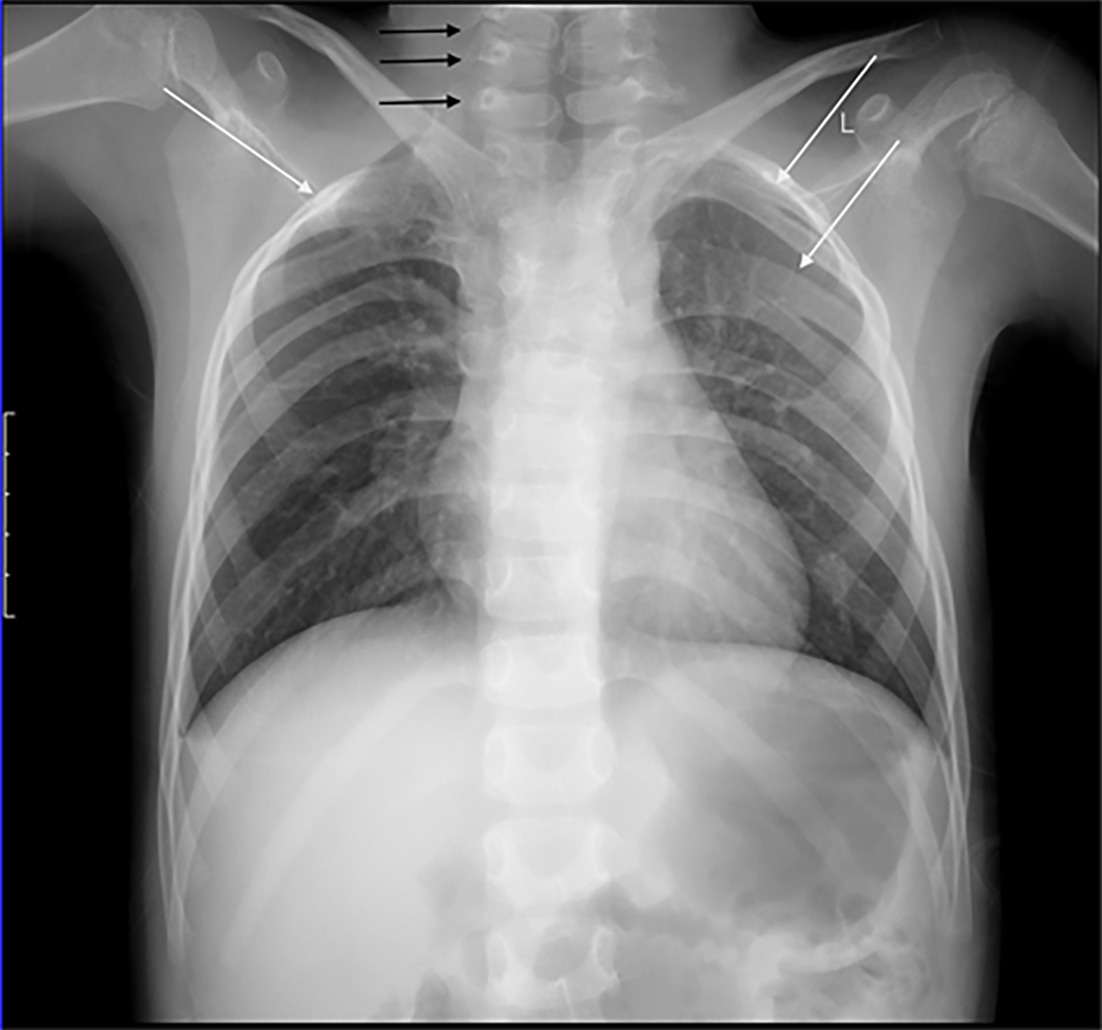
The lack of robust population screening studies performed for KFS makes it difficult to present an exact incidence for the disease;2 however, an incidence between 1 in 40,000 to 42,000 is commonly cited.2, 6, 7 A 3:2 ratio has been reported in favor of a female predominance,8 yet these statistics are difficult to determine because KFS often goes undetected and has a variety of clinical presentations.2 As such, more recent data suggest that the occurrence of KFS in the general population is much higher. A large retrospective review of cervical trauma CT scans found that the prevalence of KFS was 0.0058% (1 in 172) with a slight male predominance,6 while a prospective multicenter study of patients undergoing symptomatic cervical spinal myelopathy surgery found that 5 of 131 (~3.81%) patients had KFS.7
To assist in diagnosing KFS, classification systems based on morphology and etiology have been proposed.1 The original classification model by Maurice Klippel and Andre Feil divided KFS into 3 subtypes based on fusion extent. Advancements in genetics have greatly improved our understanding of associated molecular changes and form the basis of a new genetic subtype classification presented by Clark et al.1, 2
In the following review, we place particular emphasis on the embryology, molecular pathology, imaging diagnosis, clinical associations, and treatment of KFS.
Spinal Embryology
Understanding the embryological development of the spine is crucial for diagnosing developmental disorders of the spine, including KFS. The embryogenesis of the vertebrae begins during gastrulation.9 The paraxial mesoderm forms around the notochord and, as neurulation progresses, matures and segments into somites on both sides of the neural tube.10 Local signals cause the somites to assume a patterned and segmented appearance. From there, the dorsal somites become the dermomyotome, while the ventral somites differentiate into the sclerotome.10
The dermomyotome differentiates into muscle cells and the dermis of the skin, while the sclerotome forms the spine.11 Owing to sclerotomal rostrocaudal polarity, it undergoes resegmentation. The rostral part of the sclerotome becomes the caudal half of one vertebral body while the caudal part of the same sclerotome becomes the rostral half of subjacent vertebral body.10 Sections of adjacent sclerotomes then join to form the centrum, which stimulates the surrounding bone to develop into the vertebral body.11, 12 Concurrently, the notochord within the vertebral bodies degenerates, while the notochord remaining in the interspaces gives rise to the nuclei pulposi of the intervertebral discs.12
Pathoembryology of Vertebrae in Klippel-Feil Syndrome
KFS results from the improper segmentation of cervical vertebrae during embryogenesis, which leads to the anomalous formation of intervertebral discs.11, 12 As segmentation progresses caudally, vertebral ossification progresses rostrally.1, 13 The bidirectional nature of this process may be significant in the pathogenesis of KFS.
In KFS, the C1 vertebrae fails to develop its centrum and is therefore unable to direct the normal development of the surrounding bone. C1 remains attached to the portion of C2, which forms the odontoid process. This also prevents development of the intervertebral disc between C1 and C2.1 When the C2 vertebral body and the rostral odontoid process fail to fuse, the resulting abnormality known as os odontoideum occurs.14
The notochord also influences initial spinal cord development. During weeks 3 and 4 of gestation, the neural folds fuse dorsally to form the neural tube, a spinal cord precursor. The parallel formation of vertebrae and the spinal cord provides a possible explanation for the synchronous presence of congenital spinal cord abnormalities and vertebral bone anomalies.12
KFS is also commonly associated with a variety of other abnormalities of anomalous embryogenesis, including the Sprengel deformity of the scapula.15 At the end of the eighth week of gestation, the scapula descends into its thoracic position. Given that the scapula and midcervical vertebrae develop simultaneously, correlation between KFS and Sprengel deformity is not surprising.12
Classification
The variability of KFS has made it challenging to establish specific criteria for diagnosis and treatment. Advancements in genetics and imaging techniques have contributed to the understanding of its etiology and pathophysiology.
Klippel-Feil Classification System
Maurice Klippel and Andre Feil originally presented a case report featuring a 46-year-old French tailor with extreme discomfort in his neck secondary to fusion of the cervical and upper thoracic vertebrae.16 Based on their observations and examination of patients with similar symptoms, they proposed the first classification model for KFS ( Table 1 ). They divided the syndrome into 3 types based on the extent of vertebral fusion.1, 5, 8, 16 Type 1 is commonly associated with the classic clinical triad of KFS ( Figure 2 ).17
Klippel-Feil Classification System
| Subtypes | Vertebral Fusion |
|---|---|
| I | Massive fusion (3 or more levels) of cervical and upper thoracic vertebra with synostosis |
| II | Fusion at 1 or 2 interspaces with other cervical spine anomalies, for example, hemivertebrae, atlanto-occipital fusion |
| III | Cervical fusion associated with lower thoracic or lumbar fusion |
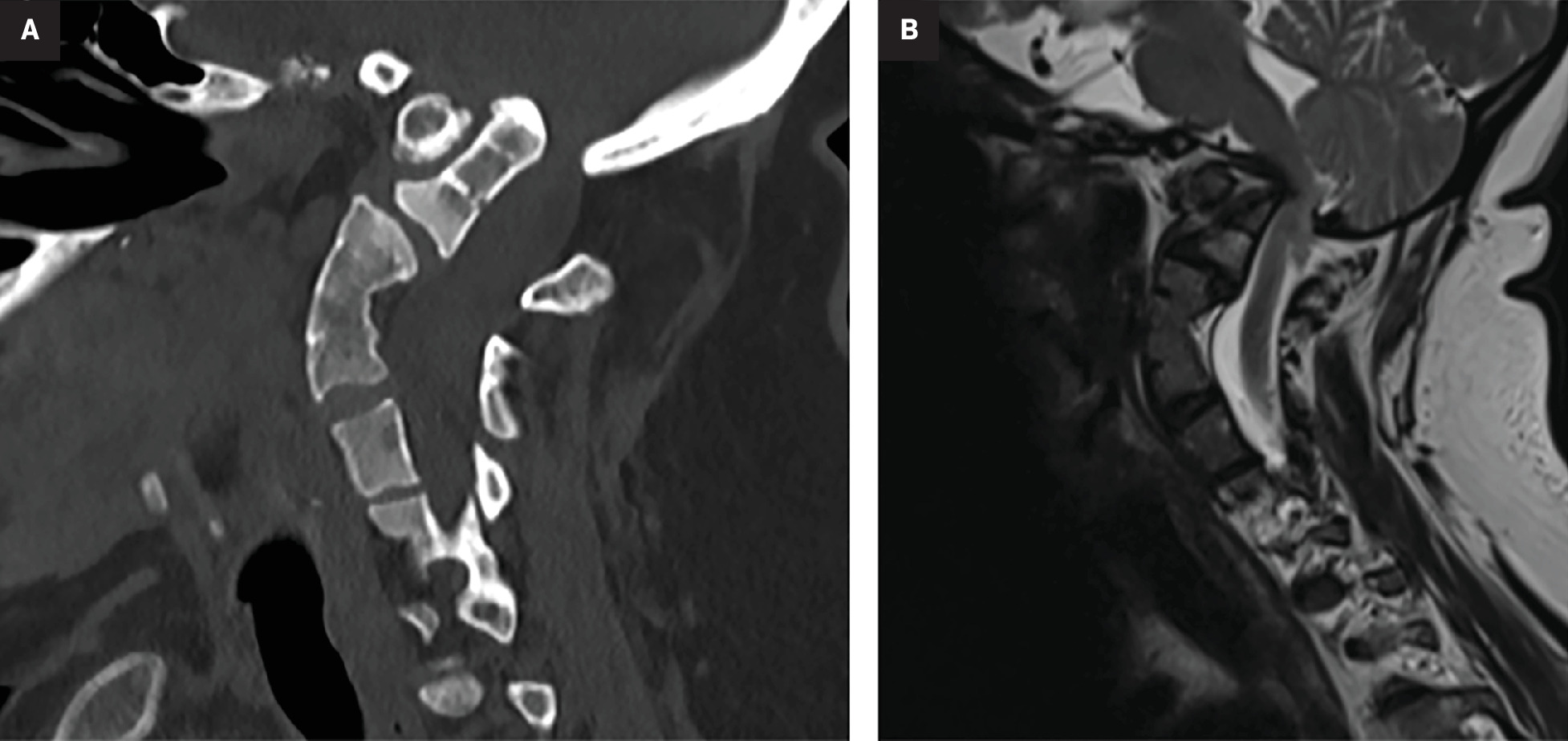
Vertebral anomalies and basilar invagination. Sagittal CT in the bone window (A) and sagittal T2-weighted MRI (B) both demonstrate multilevel segmentation abnormalities, particularly C3-C5, characteristic of Klippel-Feil syndrome. These images also reveal platybasia and basilar invagination. (B) also demonstrates the associated compression of the medulla.
Gunderson Modifications to the Klippel-Feil Classification System
The Klippel-Feil classification system of KFS has since been re-evaluated. One study by Gunderson et al18 included radiographic examination of 121 family members of patients with KFS. They demonstrated definitive cervical fusion in 11 probands and 11 relatives, with an additional 7 relatives having related cervical abnormalities. They reported that C2-3 fusion is inherited in an autosomal-dominant fashion, while C5-6 is inherited autosomal recessively, and variable cervical fusion is inherited dominantly with variable expression and penetrance. They also reported that types I and III are more severe, presenting with associated neurological and cardiac anomalies.18
Clark Classification System
In 1998, Clark proposed a classification system that considers genetic inheritance patterns in addition to morphologic presentation.1 Clark examined 3 KFS families using radiography and cytogenetic banding and found distinct variations in the timing of the postnatal vertebrae fusion and gross morphology.
Based on these differences, and to address genetic heterogeneity, they proposed a new classification system ( Table 2 ). They defined KF1 as C1 rostral fusion, which is inherited recessively. KF2 entails C2 ± 3 fusion and has dominant inheritance. KF3 usually presents as a single fusion in the ±C3 region, but also includes C5-6 fusion with recessive inheritance. Lastly, KF4 may be an X-linked vertebral fusion with a synchronous presentation of Wildervanck syndrome.1
Clark Classification of Klippel-Feil Syndrome and a Comparison with Klippel-Feil Classification System
| Class | Vertebral Fusion | Inheritance | Associated Anomalies | Overlap with Klippel-Feil Classification |
|---|---|---|---|---|
| KF1 | C1 fusion | Recessive | Short neck, cardiac, craniofacial, aural, ocular, limb | Types I-III |
| KF2 | C2-3 fusion | Dominant | Craniofacial, aural, tolaryngeal, skeletal, limb |
Types I-III |
| KF3 | Isolated fusions | Recessive or reduced penetrance | Craniofacial, facial dysmorphology | Type II |
| KF4 | Cervical vertebrae fusion | Possibly X-linked | Aural, ocular, cardiac, abducens palsy | - |
Adapted From Clark et al.1
Molecular Genetics of Spinal Development
KFS is characterized by a substantial variation in combinations of genetic and clinical presentation.1, 16, 19 Of the genes that have been studied, several show a strong association with familial KFS, as summarized in Table 3 .19 - 32
Molecular Genetics of Klippel-Feil Syndrome Development19 - 32
| Gene/Pathway | Role | Mutations |
|---|---|---|
| GDF6 | Chromosome 8 involvement, joint and vertebral development, joint fusion | Missense mutations, incomplete penetrance in carpal and tarsal fusions |
| Notch pathway | Vertebral segmental disorders, somitogenesis, embryonic patterning | Mutations in Notch1, Dll1, Dll3, Hes7, Psen1, Lfng, defects in segment borders and rostrocaudal polarity |
| PAX genes | Somite segmentation, development | Mutations linked to aniridia and Waardenburg syndrome, PAX1 mutation linked with KFS |
| HOX complexes | Vertebral development | Hoxd3 mutations cause occipitalization of atlas, Hoxd4 mutations cause supernumerary cervical vertebrae |
| BAZ1B | Association with Klippel-Feil syndrome |
Sporadic Incidence
While recent studies have shown familial inheritance, KFS has historically been understood to exhibit a largely sporadic incidence without a family history of consanguinity, potentially pointing to a significant influence of environmental or multifactorial determinants.33, 34 Tredwell examined similarities between fetal alcohol syndrome (FAS) and KFS,35 finding common physical abnormalities, such as fused cervical vertebrae, microcephaly, and thoracic cage abnormalities.
While 20% of patients with KFS had 1 cervical level involved and 50% had 4 or more involved levels, patients with FAS had rates of 53% and 5%, respectively. They also noted that alcohol was unlikely to be a contributing factor in KFS but were unable to determine a specific alternative teratogen.
Conclusion
In Part 1 our review, we have detailed the embryological development and genetic basis of KFS. This foundational understanding is essential for enhancing diagnostic accuracy and developing targeted therapeutic interventions for KFS. Part 2 of this review will focus on the clinical manifestations and diagnostic strategies for KFS.
References
Related Articles
Citation
Ritchey Z, Gunderson JR, Shaw Z, Kaddurah O, Greenhill M, King K, Mushtaq R.Klippel-Feil Syndrome Essentials, Part 1: Embryological Development and Genetic Mechanisms. Appl Radiol. 2024; (6):18 - 22.
doi:10.37549/AR-D-24-0049
December 1, 2024