Interventions for Spinal Muscular Atrophy
Images
Case Summary
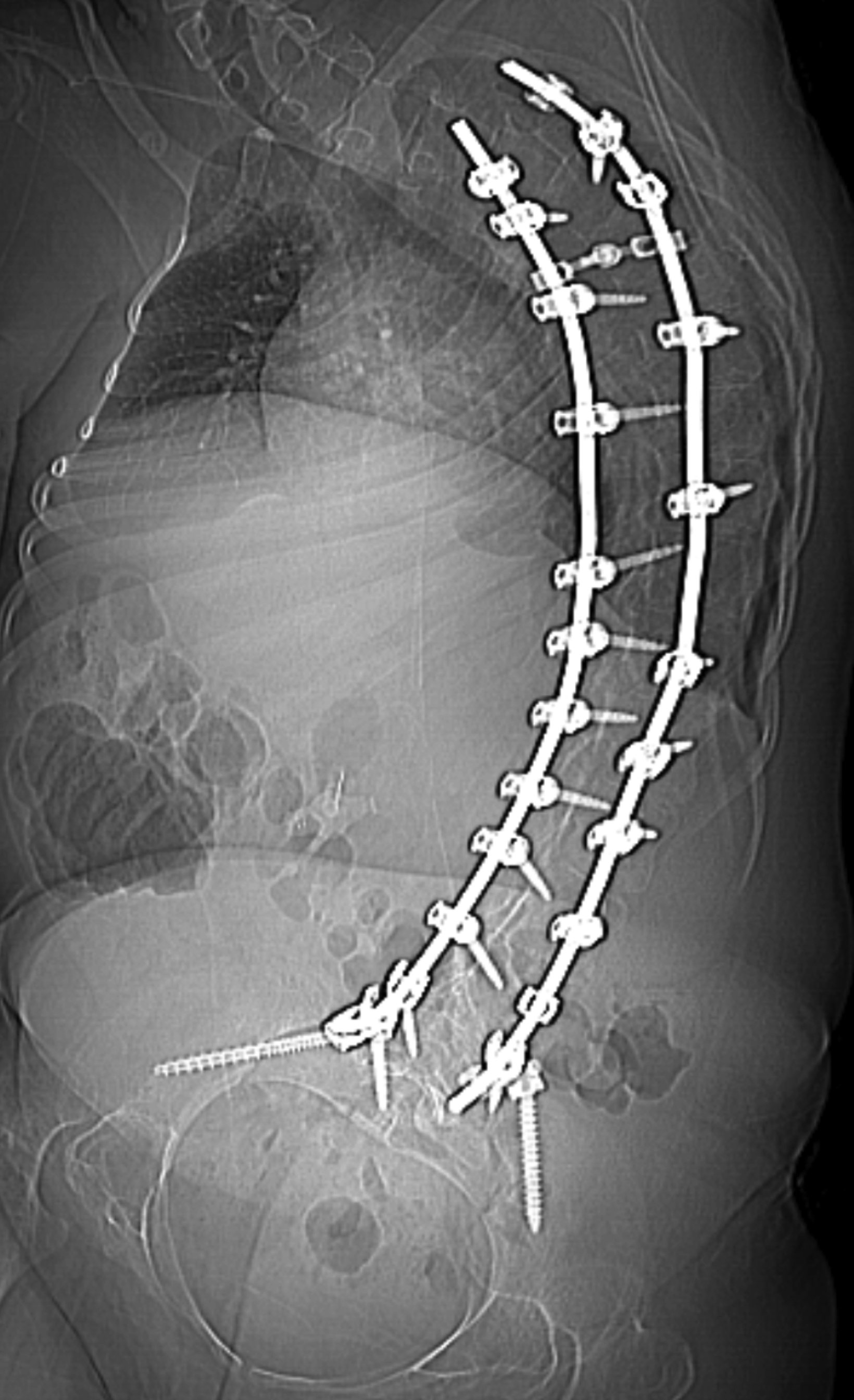
An adult with type 1 spinal muscular atrophy, with initial onset of symptoms at 5 months of age, including delayed milestones and the inability to sit independently. The diagnosis was confirmed by genetic testing. The patient had limited motor skills and had difficulties with activities necessary for independent daily living. Additionally, the patient was ventilator dependent. Spinal fixation from C4-S1 was performed for severe levoscoliosis.
Nusinersin (Spinraza, Biogen, Inc, Cambridge, Massachusetts) is an antisense oligonucleotide inhibitor that increases the amount of SMN protein, needed for normal nerve and muscle function.1 Intrathecal (IT) nusinersen was instituted at 17 years of age. Since beginning the IT therapy, hand movement and voice quality have improved, resulting in an improved quality of life.
Imaging Findings
There are no pathognomonic imaging findings diagnostic of spinal muscular atrophy. However, over time patients may develop scoliosis because of the imbalance between the flexor and extensor muscles of the trunk, typically leading to a C-shaped curvature of the spine (Figure 1). If the scoliosis is severe or progressive, spinal fusion is often necessary, as in this patient. The severity of the curvature, the vertebral rotation, and spinal fixation may make traditional approaches for IT medication administration challenging for radiologists performing these procedures.
Diagnosis
Spinal muscular atrophy (SMA). The clinical differential diagnosis is extensive and includes many conditions that present with severe hypotonia, including congenital muscular and neuropathic disorders.1
Discussion
Spinal muscular atrophy is an inherited, autosomal recessive, neuromuscular disorder most often caused by a homozygous deletion or mutation of the SMN1 gene on chromosome 5q13 with absent exon 7, resulting in absent or deficient levels of survival motor neuron (SMN) protein. A second and nearly identical gene, SMN2, produces low levels of functional SMN protein, however this fails to compensate for the loss of SMN1.2 SMN facilitates the assembly of spliceosomal small nuclear ribonucleoprotein (snRNP) particles, which are thought to function in motor neuron growth and neuromuscular maturation.3 Over time, SMA results in the degeneration of anterior horn cells in the spinal cord with associated destruction of alpha motor units in lower motor neurons.
SMA presents with varying clinical phenotypes and is classified into five main subtypes based on severity and age of onset. All are characterized by progressive muscle weakness, reduced tone, and atrophy. However, cognition is not affected. Type 0 is congenital SMA, which presents early in neonatal life as severe hypotonia, early respiratory failure, and severe weakness, with death occurring at birth or within the first month of life. Type 1, also known as Werdnig-Hoffman disease, presents in the first 6 months of life with limited head control, hypotonia, areflexia, weakness of intercostal muscles, swallowing difficulties, and tongue fasciculations.
Type 2, Dubowitz disease, is of intermediate severity, presenting at 6-18 months of age. Children with SMA 2 are able to sit but have hypotonia, areflexia, progressive scoliosis, and restrictive lung disease. Mortality is most commonly due to respiratory compromise, but 70% of patients survive until age 25.
Type 3, Kugelberg-Welander disease, is mild SMA, presenting after 18 months of age with more progressive proximal weakness in the legs than in the arms. Type 3 patients are ambulatory and typically do not have restrictive lung disease. Additionally, their life expectancy is not affected.
Finally, type 4 is adult-onset SMA. It has the mildest phenotype and presents in patients older than 21 years, with progressive, mild proximal weakness typically not impairing ambulation or affecting life expectancy.4
If clinical history and physical exam findings raise the suspicion for SMA, the diagnosis can be confirmed by molecular genetic testing to detect homozygous exon 7 deletion in the SMN1 gene, which is 100% specific for diagnosis. Around 5% of patients with SMA are compound heterozygotes with a single SMN1 deletion and a frameshift, nonsense, or missense mutation in the other SMN1 copy. SMN2 differs from SMN1 by a single nucleotide that disrupts a splice enhancer in exon 7, producing an unstable protein that is unable to compensate for the loss of SMN1. However, approximately 10-20% fully functional, full-length transcripts are generated from SMN2.
There are zero to eight SMN2 copies in the genome, which is inversely correlated with disease severity. A higher number of SMN2 copies is often able to produce a milder type 2 or type 3 phenotype of SMA. In the normal population, the SMN2 copy number varies from zero to three, with 15% of normal individuals having no SMN2. An analysis of 625 unrelated Spanish patients with SMA showed that the majority of individuals with type 1 SMA had one or two SMN2 copies;, most of those with type 2 SMA had three gene copies; and those with type 3 had three or four SMN2 copies.5
Therapeutic approaches are multi-fold, including splicing modification of SMN2 via antisense oligonucleotides (ASO), small molecules which modulate SMN2 gene splicing, and SMN1 gene replacement therapy. Nusinersen, the first molecular drug to treat SMA, is an intrathecally administered ASO which promotes inclusion of exon 7 in mRNA transcripts of SMN2, resulting in translation of a higher level of fully functional SMN protein in liver, kidney, skeletal muscle, and central nervous system tissues. SMA patients have experienced significant improvement in motor function and extension in life expectancy after receiving nusinersen.7
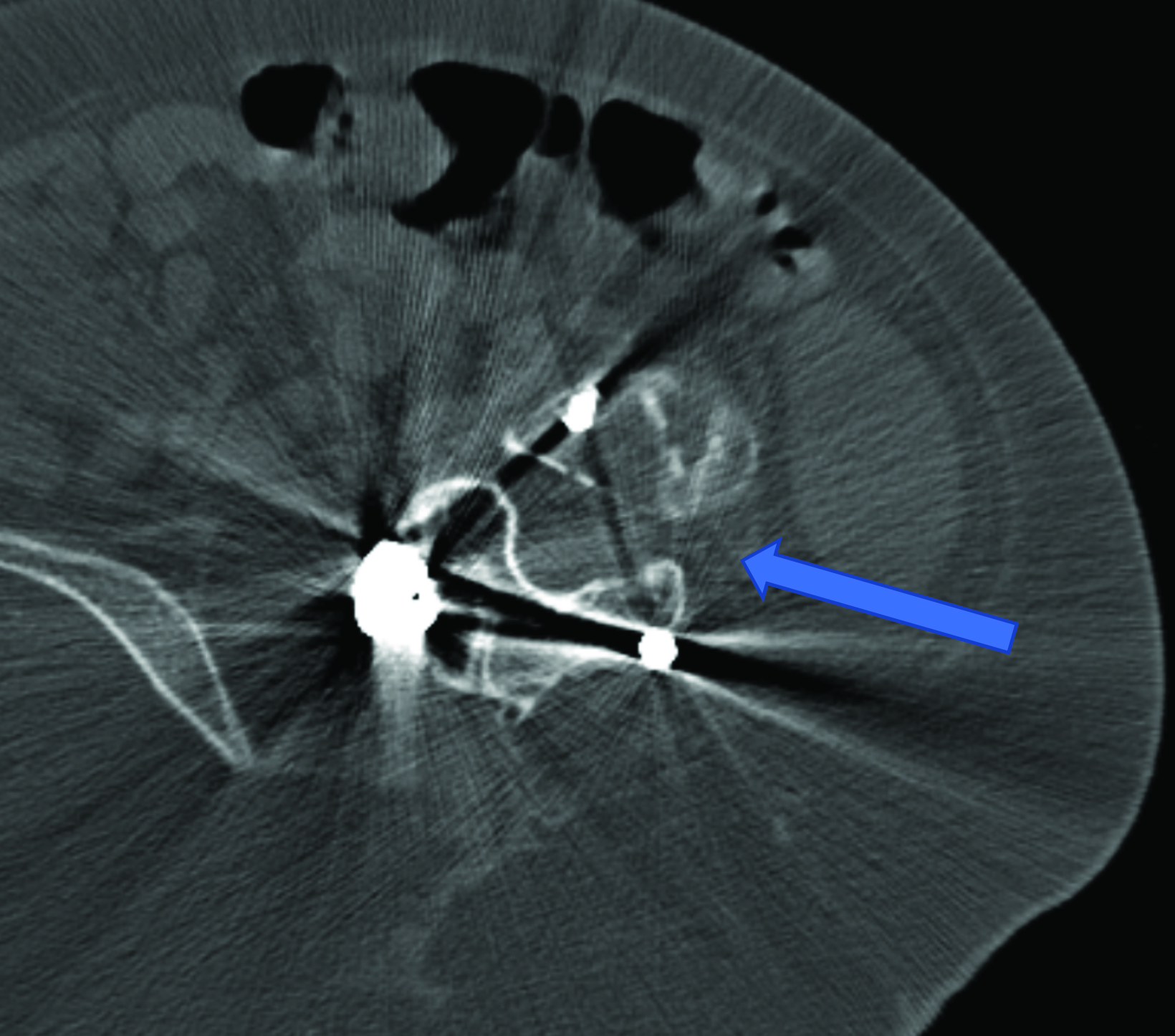
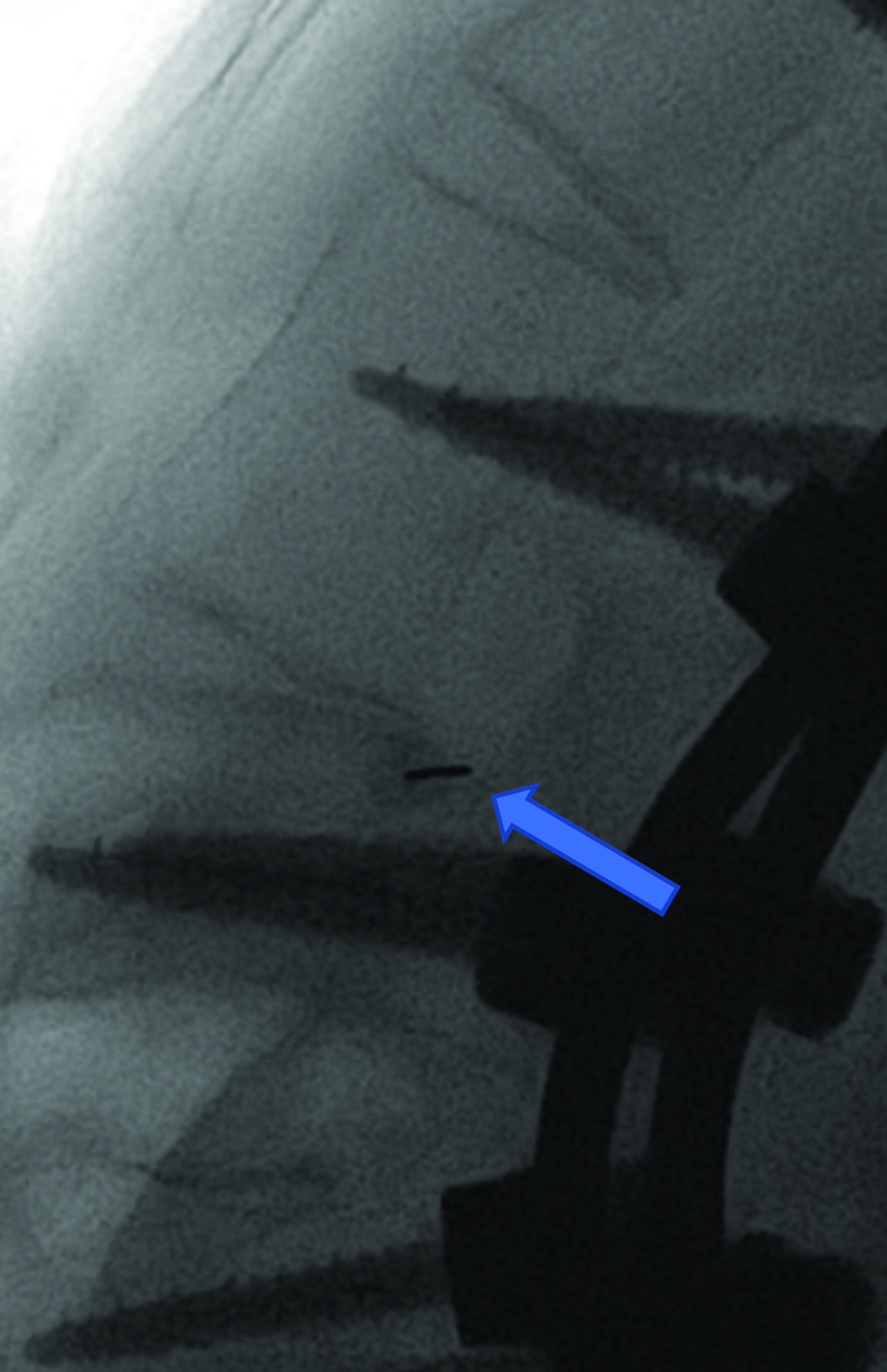
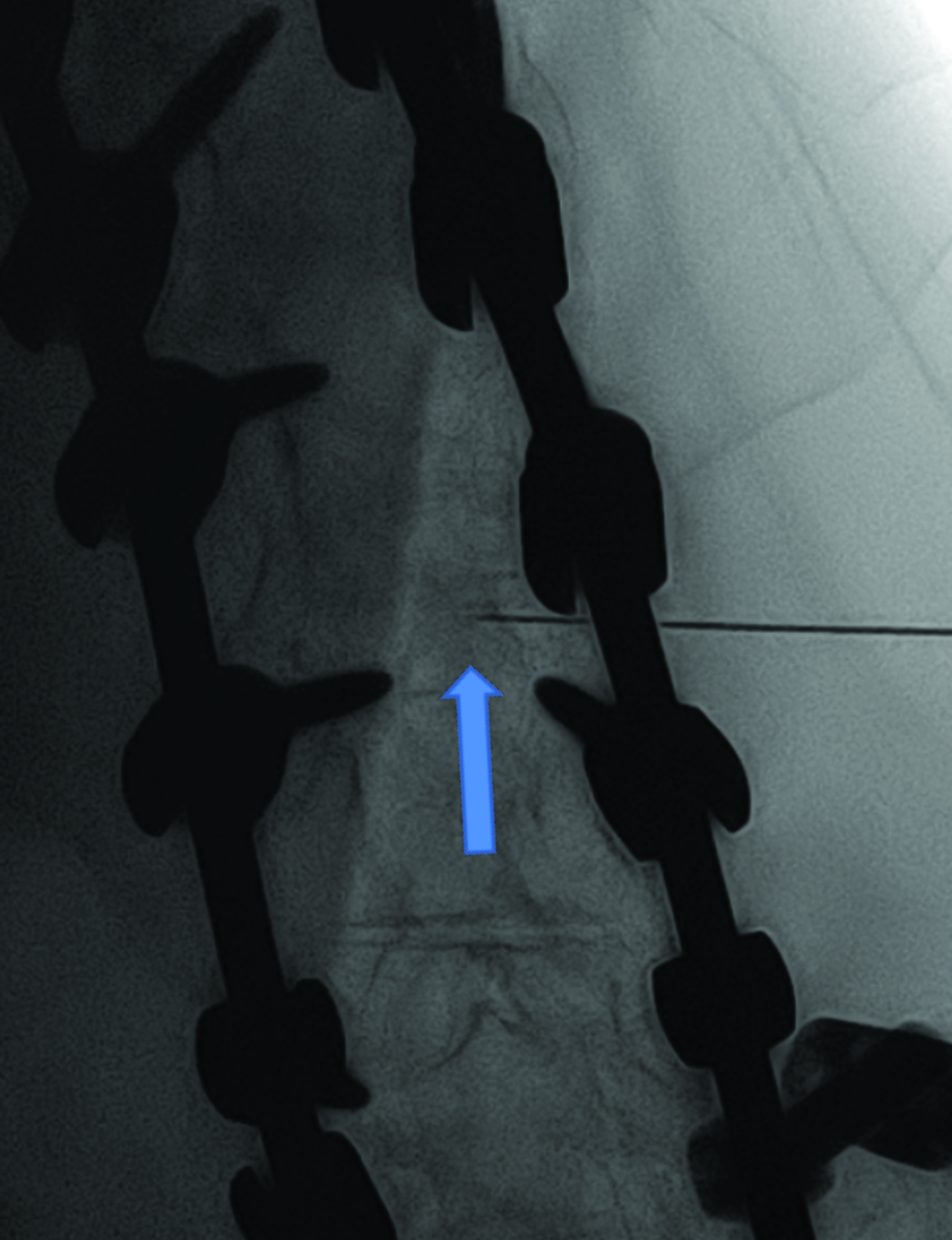
Intrathecal access in patients with SMA may be challenging based on whether patients have simple or complex spinal anatomy (neuromuscular scoliosis, spinal fusion, and/or instrumentation, Figure 2). Several approaches can be used to facilitate drug injection into the subarachnoid space. In children with uncomplicated spines, a midline lumbar puncture is performed. In those with complex spines, lumbar transforaminal or C1-2 approaches may be required. Although either approach can deliver IT therapy, it is believed that the lumbar approach provides a higher drug concentration to the target lower motor neurons.
The authors prefer the transforaminal approach, as it is reported to be safer and technically easier than the C1-2 approach.8,9 Multiple reviews have shown that the complex anatomy commonly present in patients with SMA usually requires preprocedural imaging for careful planning, as well as intraprocedural guidance to access the subarachnoid space via a transforaminal approach.8,9
A newer, orally administered small molecule called risdiplam (Evrysdi, Genentech) has also been developed to treat SMA. This drug modulates SMN2 gene splicing by binding two pre-mRNA splice sites in exon 7, increasing levels of full-length SMN mRNA and protein. Risdiplam crosses the blood-brain barrier, achieving systemic distribution with suitable half-life and predictable pharmacokinetics.10
Onasemnogene abeparvovec (Zolgensma, Novartis) is an SMN1 gene replacement therapy, which uses a non-replicating, adeno-associated virus capsid carrying SMN1 complementary recombinant DNA to efficiently deliver wild-type SMN1 to motor neuron cells. The drug cannot reverse any pre-existing damage to motor neurons and is currently approved for children under age 2 years.11 To achieve the best results it is important to give the drug early in the disease course before there is injury to the motor neurons.
Conclusion
Spinal muscular atrophy is an inherited neuromuscular disorder characterized by progressive muscle weakness. It has varying clinical severity ranging from mild proximal muscle weakness to death from restrictive lung disease. The diagnosis is confirmed by genetic testing. Treatment includes the SMN2 splicing modulator nusinersen, facilitated by interventional radiologists. This may be challenging to administer intrathecally as patients develop scoliosis over time, making midline or paramedian lumbar punctures more challenging. Patients with complex spinal anatomy may benefit from pre- and intraprocedural imaging to allow for a transforaminal approach to the thecal sac.
References
Citation
RB DJT, CM S, AJ T. Interventions for Spinal Muscular Atrophy. Appl Radiol. 2023;(3):53-55.
May 5, 2023