Congenital Pulmonary Airway Malformation
Images
Case Summary
A pregnant patient presented at 35 weeks ’ gestation for further evaluation of a previously diagnosed fetal congenital pulmonary airway malformation (CPAM). The baby was asymptomatic at near-term delivery and discharged home. At 9 months of age a left-sided thoracoscopy and left lower lobectomy for resection of the CPAM was performed.
Imaging Findings
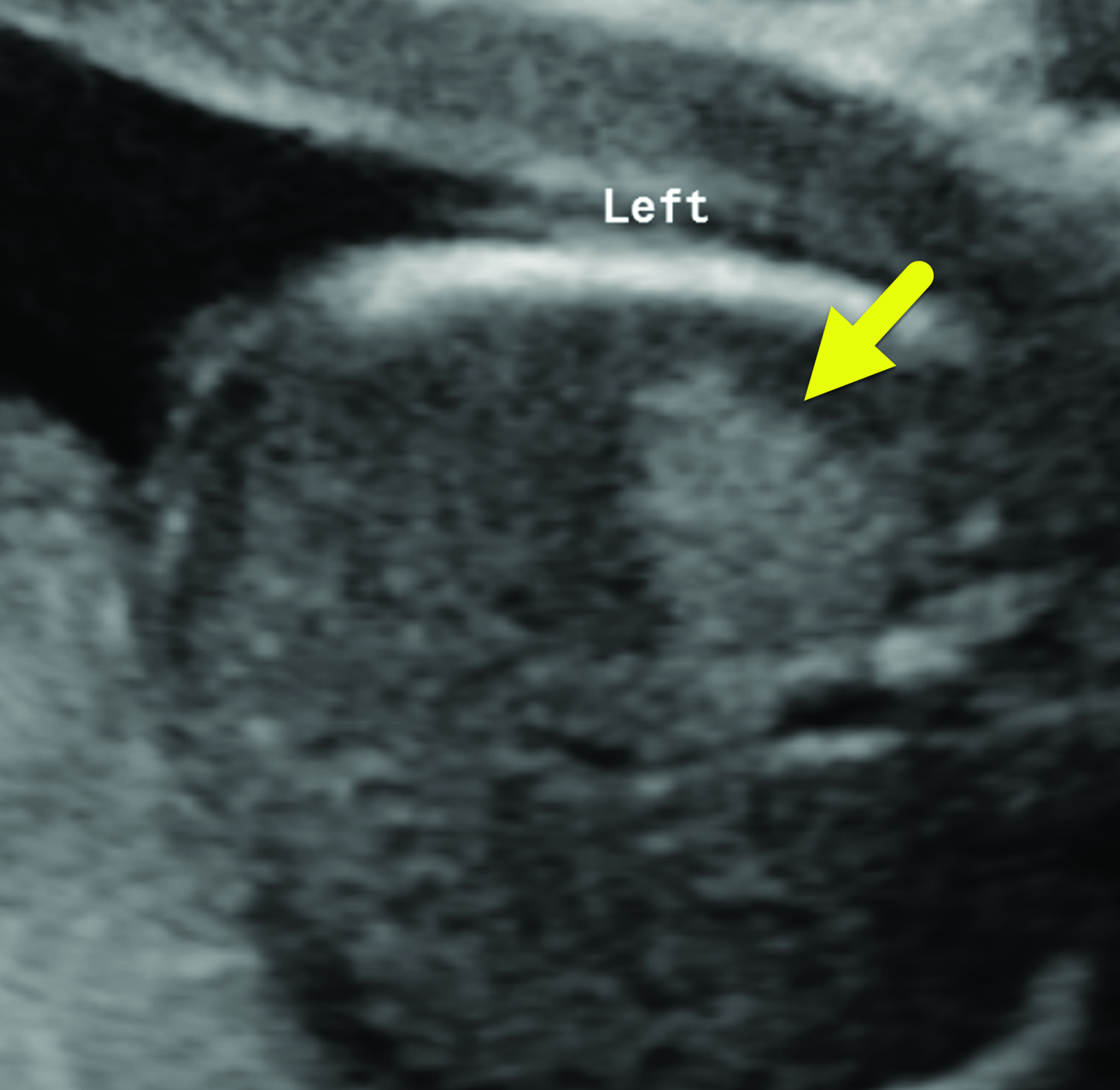
Prenatal ultrasound (Figure 1) showed a 2.7 × 1.9 × 1.2 cm left-sided mass with a single cyst. The CPAM volume ratio (CVR) was 0.1 and had increased in size from a prior study, when it was 0.06. This clinically important ratio can help predict which fetuses will be more likely to develop fetal hydrops, a CVR < 1.6 being associated with a lower likelihood.
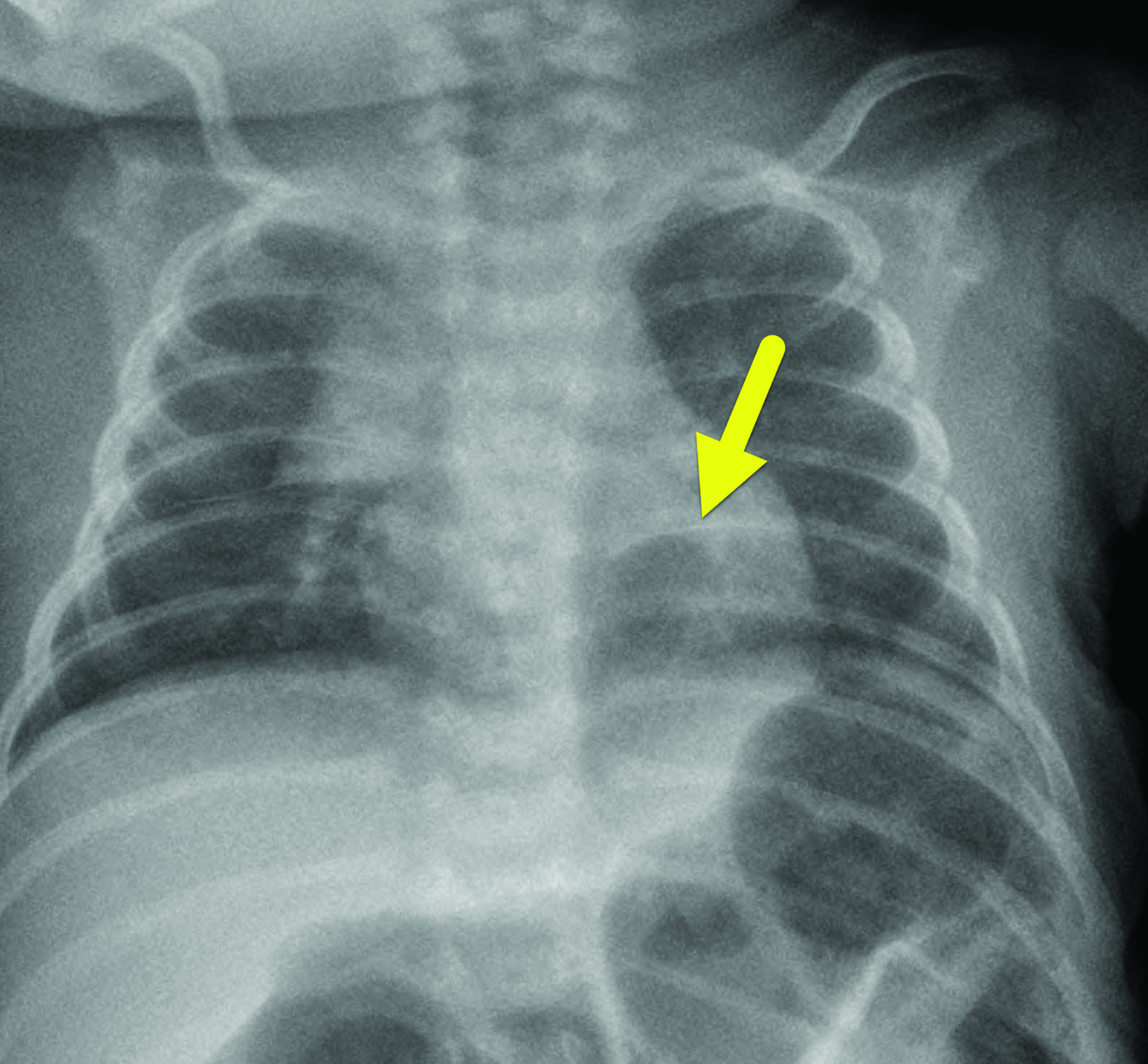
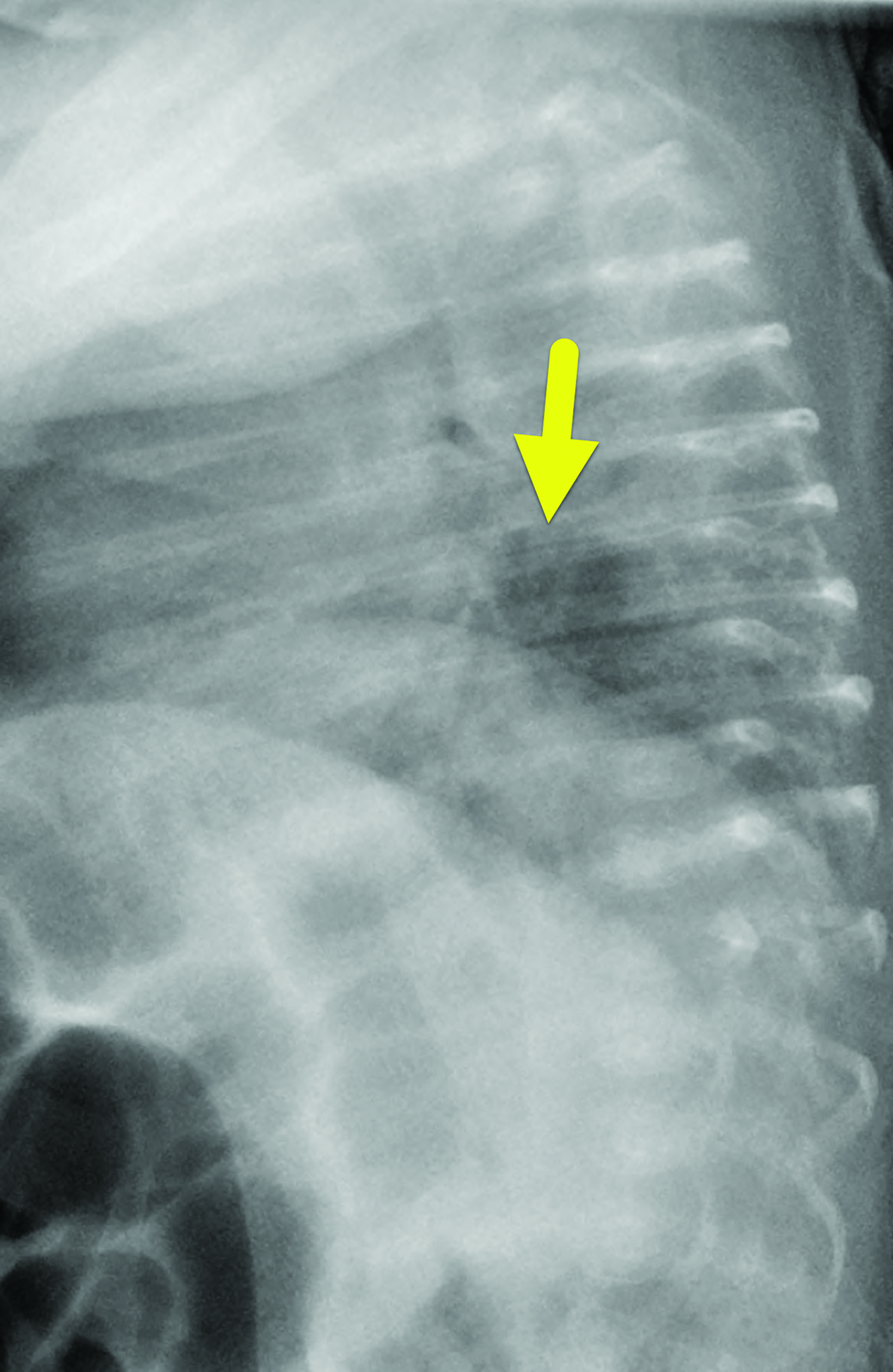
Chest radiography (Figure 2) performed at 1 month of age showed a relative lucency in the posterior aspect of the left lower lobe. There was no pneumothorax, pleural effusion, or mediastinal shift.
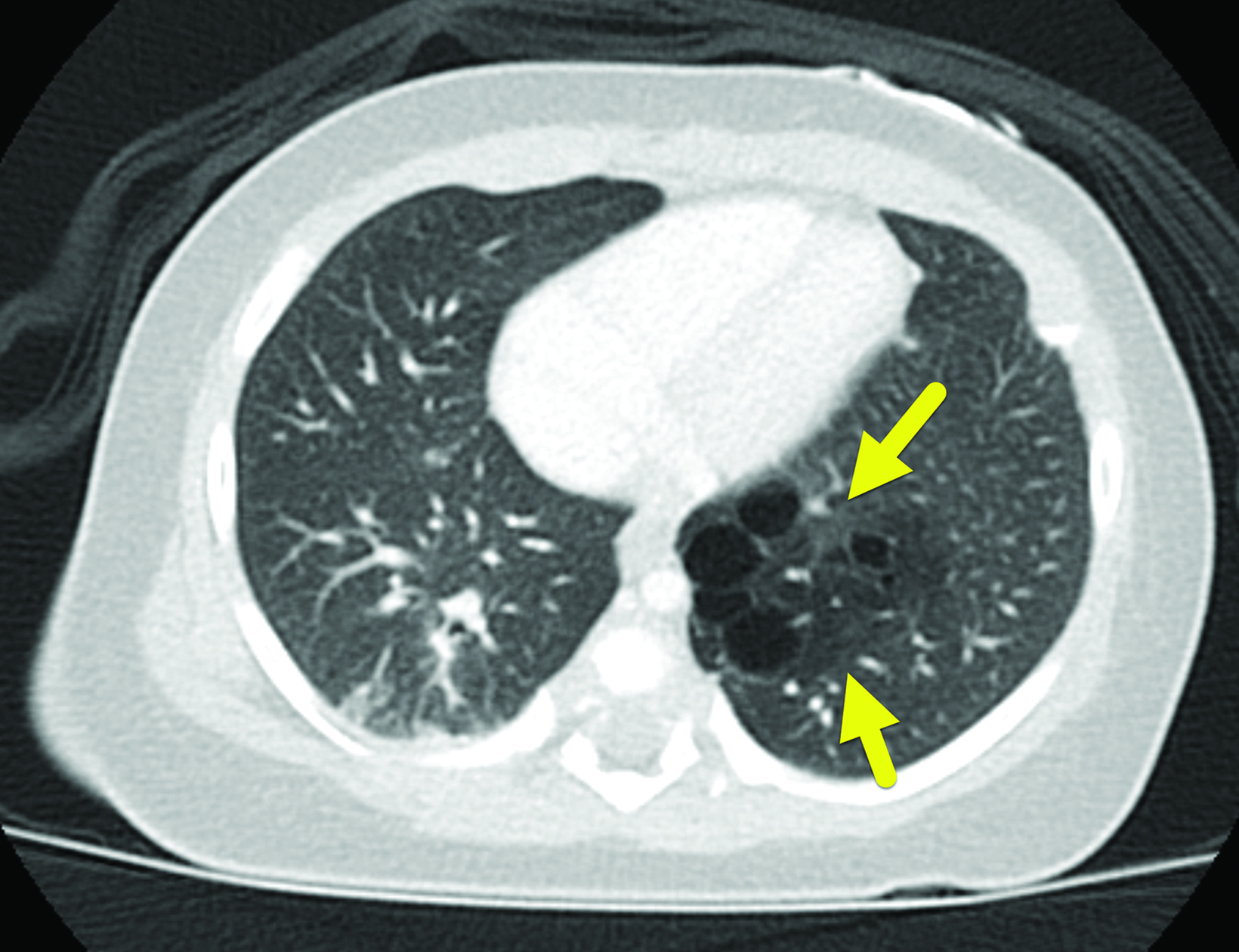
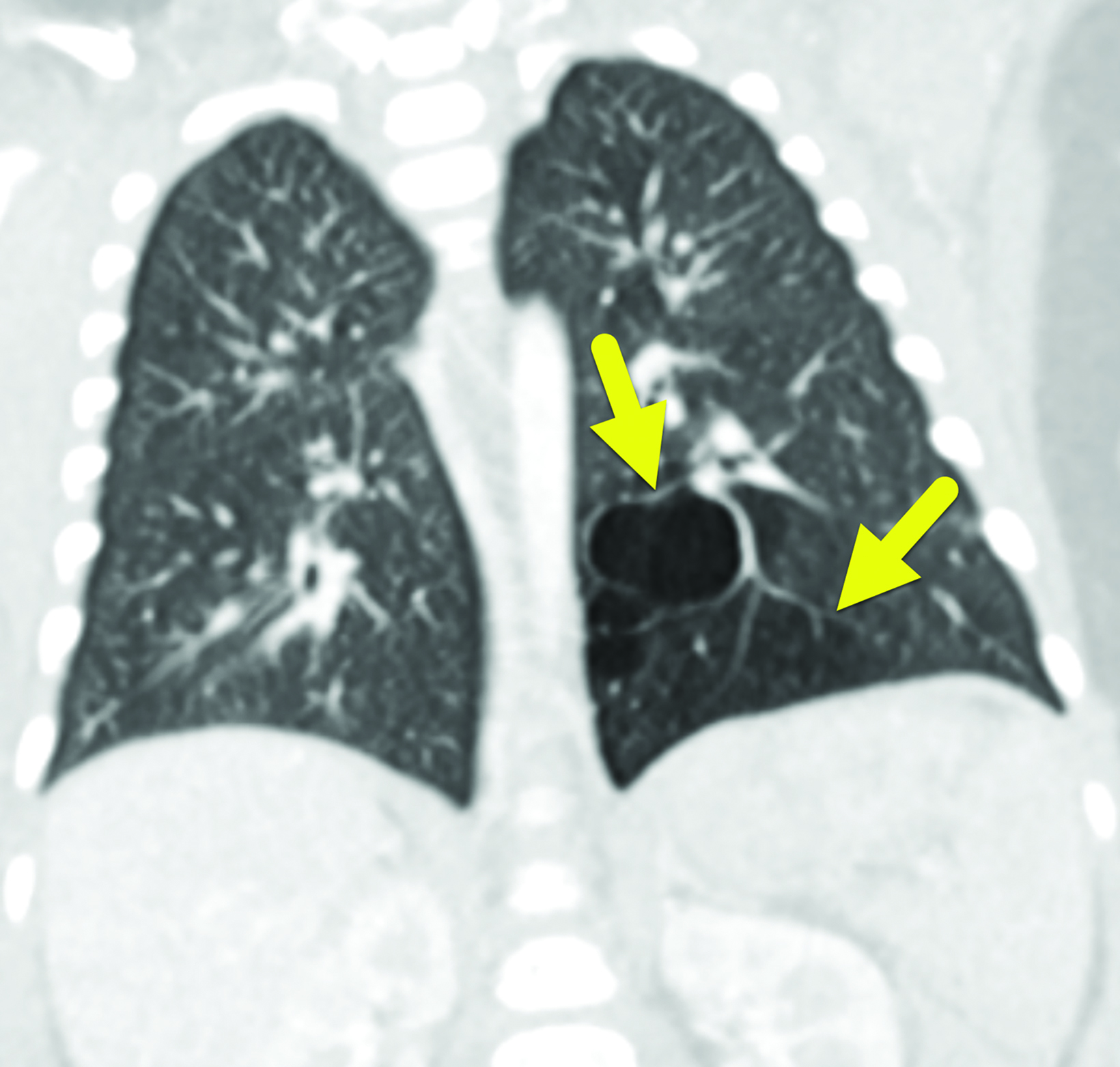
Contrast-enhanced chest CT (Figure 3) performed at 5 months showed a 3.6 × 3.3 × 3.8 cm lesion in the left lower lobe that contained multiple air-filled cysts within a soft-tissue mass and an associated atretic bronchus. There was no systemic arterial supply.
Diagnosis
Differential diagnoses include pulmonary sequestration, congenital lobar overinflation, and a bronchogenic cyst.1
Discussion
Congenital pulmonary airway malformation is the most common congenital lung lesion of the lower respiratory tract. It was formerly known as congenital cystic adenomatoid malformation (CCAM).1 The name was changed to better conform to the histologic variety.
CPAM occurs with a reported incidence of 1 in 10,000-35,000 births. The condition may be diagnosed in utero, in the neonatal period or later, or while the patient is undergoing thoracic imaging for an unrelated reason.2
Five Types of CPAMs
CPAMs can be classified histologically or by antenatal sonographic appearance. The Stocker classification is based on histologic features and specifies five types of lesions. Each type is derived from a component of the bronchial tree which informs the histopathology. The often-fatal type 0, which originates in the trachea or bronchi, is characterized by cartilage, smooth muscle, glands, and mesenchyme. Type 1 CPAMs stem from the bronchi and/or bronchioles and is the most common form of of the condition. It is characterized by large pulmonary cysts lined with mucus cells and pseudo-stratified columnar epithelium.
Type 2 CPAMs consist of multiple small cysts that originate from bronchiolar areas. These lesions are often associated with renal, cardiovascular, and skeletal abnormalities. Type 3 CPAMs are bulky, solid lesions that can cause mediastinal shift. They also origi- nate from the bronchiolar regions. Finally, type 4 CPAMs originate from acinar lung tissue and contain variably sized cysts with type 1 and type 2 alveolar cells.1-4
Diagnosis
CPAM is frequently diagnosed with fetal ultrasound during the second trimester; the Adzick classification is used to sonographically categorize the lesions.1 In this classification, type 1 lesions are macrocystic with one or more cysts > 5 mm and type 2 (micro- cystic) lesions contain cysts < 5 mm. The larger cysts in type 1 CPAM appear cystic with hypoechoic fluid. Type 2 lesions are solid-appearing and echogenic.2,5
Pulmonary sequestration is typically part of the differential diagnosis for an echogenic fetal lung lesion. Pulmonary sequestration differs from CPAM in that it reflects an isolated portion of lung that receives its blood supply from an abnormal systemic artery and does not communicate with the bronchial tree. CPAM, in contrast, maintains communication with the bronchial tree and has a normal pulmonary blood supply. Fetal ultrasound with color Doppler evaluation can help distinguish these vessels.
Other entities in the differential diagnosis of an echogenic fetal lung lesion include congenital lobar overinflation and bronchogenic cysts. Congenital lobar overinflation creates mass effect and can compress neighboring structures. It is caused by an intrinsic bronchial epithelial defect or external obstruction that leads to air trapping. Bronchogenic cysts are unilocular cysts filled with fluid or mucus and lined by pseudostratified ciliated columnar respiratory epithelium and hyaline cartilage.2
CPAM identified in utero may spontaneously resolve, regress, or progress. A retrospective review of 100 fetuses with congenital lung lesions found that vanishing lung lesions were more likely to occur with microcystic lesions or in those with a low CVR.6 Even when a lesion has resolved in utero, postnatal imaging is recommended to confirm resolution and characterize any sonographically occult residual.
Potential associated abnormalities are assessed on prenatal imaging; these include mediastinal shift, fetal hydrops, hydramnios, and fetal demise. The CVR was developed by Crombleholme, et al, to assess risk for CPAM-related complications. It represents the ratio between the prenatal sonographic estimate of CPAM volume (length × height × width × 0.52) and head circumference. A value greater than 1.6 is associated with an 80% higher risk of fetal hydrops.
Follow-up fetal ultrasound frequency is based on the CVR.1,2,8 Postnatal imaging begins with a chest X-ray, repeated six months later to assess for lesion stability. On chest radiography a CPAM typically appears as a lucent mass with varying areas of radio-opacity depending on CPAM type. CT angiography (CTA) will characterize the lesion, facilitate surgical planning and identify the arterial supply (pulmonary vs systemic).1,7 Specimen analysis after surgical resection is the most reliable method for histological confirmation and typing.1,2
If not diagnosed antenatally, CPAM is usually diagnosed following chest imaging for unrelated reasons or for pulmonary symptoms.
Treatment Options
Treatment can be considered antenatally and postnatally and should be guided by the presence of complications. Ideal evaluation for antenatal treatment includes fetal MRI for lesion and vascular characterization and echocardiography to evaluate for valvular regurgitation or impaired ventricular function.1 Treatment options include maternal steroids, percutaneous laser ablation of the lesion, minimally invasive and/or open fetal surgery.1,2
For microcystic lesions with hydrops, maternal administration of steroids is associated with fewer complications than open surgery. Macrocystic CPAM with hydrops can be treated with fetal thoracocentesis; however, thoracoamniotic shunting is the most effective treatment, as it prevents re-accumulation of fluid. Mediastinal shift resulting from large lung lesions can be addressed with ex-utero intrapartum treatment procedure if fetal lung development is sufficient.1
Postnatal treatment of CPAM is surgical (thoracoscopic or open). There is clear benefit for surgical resection in symptomatic patients; however, the optimal management of asymptomatic lesions is less clear, as the natural progression of postnatal CPAM is not fully understood. Symptomatic patients may present with lesion infection, respiratory distress, pneumothorax, hemorrhage, feeding difficulties, acute hypoxic events, and malignant transformation, although malignancy is relatively rare.1
Resection of asymptomatic lesions is often recommended to prevent complications. Additionally, type 4 CPAM cannot be differentiated from pleuro-pulmonary blastoma (PPB) on imaging.1,9 While PPB is rare, it is an aggressive cancer. Thus, the potential for prevention can be a significant benefit to electively removing an asymptomatic type 4 lesion.1 Other associated malignancies include bronchoalveolar carcinoma and rhabdomyosarcoma.2 Another potential advantage of electively resecting an asymptomatic CPAM is a lower rate of surgical complications compared with emergent surgery for symptomatic patients. Observation is an option for asymptomatic patients.1,2,9,10
Conclusion
Congenital pulmonary airway malformations form from abnormal airway branching and is the most common congenital lung lesion. Prenatal ultrasound has increased antenatal diagnosis and improved prognosis. CPAM can be classified by its histopathological appearance or sonographic characteristics.
Postnatal diagnosis is typically made after clinical presentation of infection or respiratory distress. Chest radiography and CTA can characterize CPAM. However, diagnosis is most reliably made with specimen analysis, which can differentiate CPAM from rare but aggressive lesions.
References
Citation
E L, RB T, CM S, AJ T.Congenital Pulmonary Airway Malformation. Appl Radiol. 2023; (5):34-37.
September 12, 2023