Pheochromocytoma
By Saeed OB, Towbin RT, Towbin AJ
CASE SUMMARY
A previously healthy 15-year-old was evaluated for secondary hypertension with severe hypertension (150/77), syncope, occipital headache, episodic flushing, exertional chest pain, and diarrhea for approximately 1 year.
IMAGING FINDINGS
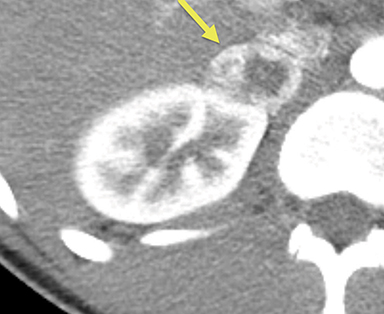
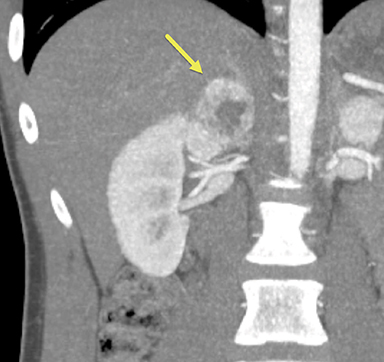
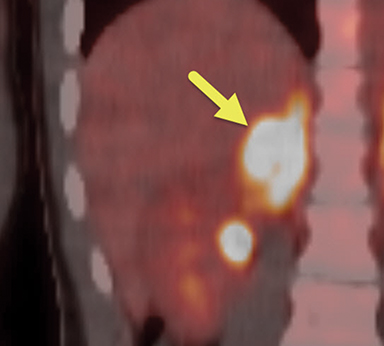
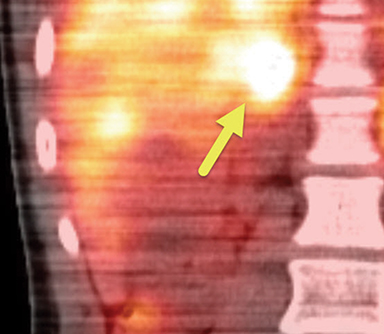
Initial renal ultrasound with Doppler interrogation, electrocardiogram, and echocardiogram were normal. A CT scan of the abdomen showed a heterogeneous right adrenal mass with peripheral contrast enhancement (Figure 1). The left adrenal gland and kidneys were normal. Fluorodeoxyglucose (FDG)-positron emission tomography (PET) whole body scan showed an FDG-avid right adrenal mass, with relative central photopenia (Figure 2). No nodal or distant metastatic disease was identified. MIBG of the adrenal gland showed uptake within the right adrenal mass (Figure 3).
Laboratory testing showed urinary normetanephrine levels of 1400 mcg/24 hours (Upper limit of normal (ULN): 450 mcg/24 hours), urinary norepinephrine 400 mcg/24 hours (ULN: 105 mcg/24 hours), plasma norepinephrine 10047 pg/mL (ULN for an 11-15-year-old: 287 pg/mL), and plasma dopamine 73 pg/mL (ULN:<60pg/mL).
DIAGNOSIS
Pheochromocytoma. The differential diagnosis for a right adrenal mass in an adolescent includes adrenal adenoma, pheochromocytoma, and adrenocortical carcinoma. Neuroblastic tumors, including neuroblastomas, are the most common adrenal tumors of childhood. However, they are not common in adolescence.1
DISCUSSION
Pheochromocytoma and paragangliomas are rare neuroendocrine tumors that arise from chromaffin cells. Paragangliomas form in sympathetic tissue in the adrenal gland or near blood vessels, and from parasympathetic tissue in the head and neck. Paraganglioma that forms in the adrenal gland is known as pheochromocytoma.2 Pheochromocytoma was first described by Frankel (1886)3 and the first successful removal of a pheochromocytoma was performed by Roux in 1926.4
Pheochromocytomas are present in 0.05 – 0.1% of patients with sustained hypertension.5 While pheochromocytomas can occur at any age, they most commonly occur in middle-aged men and women. Pediatric patients account for 10% of all patients diagnosed with the tumor.6 Children diagnosed with pheochromocytoma are more likely to be male (M:F = 2:1) and have a family history of the disease.7 Fewer than 10% of pediatric pheochromocytomas are malignant and pediatric patients have a mean survival rate of 73% at 3 years.5
Classically, patients with pheochromocytoma present with brief recurrent episodes of hypertension, palpitations, headaches, and sweating; an acute attack may consist of pallor, nausea, and panic attacks.8 Other nonspecific signs include flushing, tiredness, or weight loss.7 Attacks may be precipitated by palpation of the tumor, postural changes, exertion, anxiety, and food or beverages containing tyramine (cheese, beer, wine)2 The symptoms are caused by production of excess catecholamines.
CT is the modality of choice for pheochromocytoma screening, as it may also be used to identify renovascular causes of hypertension.9 On CT, a pheochromocytoma has a heterogeneous appearance with small cystic areas. Calcifications or hemorrhage may also be seen. On dual-phase contrast-enhanced CT, pheochromocytomas can be distinguished from other adrenal masses due to higher intensity during the arterial phase, with enhancement levels greater than 10 HU and washout less than 50%.2
MRI can also be utilized for initial diagnosis or follow-up imaging. Classically, pheochromocytomas are “lightbulb bright” on T2 MRI.2 Furthermore, the signal intensity of a hemorrhagic tumor is predominantly high on T1 images.10 Smaller, solid pheochromocytomas can be distinguished from adrenal adenomas by chemical shift imaging; unlike adenomas, pheochromocytomas contain no intracellular lipids. Thus, they show no signal changes in out-of-phase and in-phase images. Additionally, diffusion weighted imaging and apparent diffusion coefficient values are significantly higher in pheochromocytomas compared to adenomas and metastases.11
Functional imaging allows for determination of disease severity and is recommended for patients with metastatic paragangliomas or increased risk for metastasis (large tumor size).2 123I metaiodobenzylguanidine (MIBG) scintigraphy has a high sensitivity for pheochromocytoma and is useful in diagnosing the primary tumor, as well as diagnosing and evaluating metastatic disease.2,9
Pheochromocytomas periodically secrete excess catecholamines, including epinephrine, norepinephrine, and dopamine. Because of the periodic nature of the release, a one-time estimation of plasma or urinary catecholamines may be insufficient. The metabolites of catecholamines (epinephrine is metabolized to metanephrine, and norepinephrine is metabolized to normetanephrine), however, are constantly in circulation and can be detected in the plasma and urine.12 Thus, the most sensitive tests as recommended by the U.S. Endocrine Society are measurements of fractionated metanephrines in urine or plasma.9
Most pheochromocytomas are benign, but approximately 10% are malignant and can metastasize. Paragangliomas that are more likely to metastasize tend to be larger than 5 cm, located outside the adrenal glands, diagnosed in younger patients, and contain an SDHB mutation.13 Paragangliomas and pheochromocytomas tend to metastasize to the lymph nodes, liver, bones, and lungs.13
Up to 25% of pheochromocytomas are hereditary.14 Six pheochromocytoma-associated syndromes have been described in the literature including neurofibromatosis type 1, multiple endocrine neoplasia type 2, von Hippel-Lindau Syndrome (VHL), and paraganglioma syndromes types 1, 3 and 4.14 Pheochromocytomas occurring in the setting of VHL or paraganglioma syndromes type 1 or 4 tend to occur in younger patients.15
Pediatric pheochromocytoma is effectively treated with surgical removal.7 The preferred surgical treatment for pheochromocytomas includes laparoscopic resection with adrenal cortical sparing; between 15 and 30% of the adrenal gland is needed to preserve function.2,5,8 One pediatric study reported an overall survival rate of 100% of patients with benign disease, and 78%, 62%, and 31% at 5 and 10 years and 15 years, respectively, of those with malignant tumors.16
To reduce the risk of complications during surgery, patients are preoperatively managed with alpha blockade and beta blockade with a blood pressure reduction goal of <50th percentile for age and height.7 These medications are given to prevent severe hypertensive crises seen with alpha receptor stimulation and to prevent reflex tachycardia from alpha blockade. Patients are also instructed to consume a high sodium (6-10 grams) diet and have high fluid intake to prevent hypotension.7
Long-term follow-up is necessary due to the risk of recurrence. The rate of paraganglioma recurrence after resection ranges from 12% to 38%.7,17 Recurrence increases with time, from 25% at 9 years to 50% at 31 years.17 Recurrences are more common in patients with germline mutations.17
CONCLUSION
Pheochromocytoma is a rare neuroendocrine tumor of the adrenal gland that classically causes paroxysms of hypertension, headaches, palpitations, and sweating. Most cases are benign and are characterized by excess production of catecholamines. While pheochromocytomas most commonly occur in the 3rd to 5th decade of life, they can occur at any age. Children diagnosed with pheochromocytoma are more likely to have an underlying syndrome such as VHL. Diagnosis is often initially made via CT or MRI and is confirmed with plasma and urinary metanephrines. Management is with resection of the tumor with high survival rates for benign disease. Long-term follow-up is necessary due to high rates of recurrence.
REFERENCES
- McHugh K. Renal and adrenal tumours in children. Cancer Imaging. 2007;7(1):41-51. Published 2007 Mar 5. doi:10.1102/1470-7330.2007.0007
- Pacak K, Tella SH. Pheochromocytoma and Paraganglioma. In: Feingold KR, Anawalt B, Boyce A, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000. http://www.ncbi.nlm.nih.gov/books/NBK481899/. Accessed December 19, 2019.
- Frankel F. Ein Fall von doppelseitigem, vollig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veranderungen am Circulationsapparat und Retinitis. Arch Pathol Anat Physiol Klin Med. 1886; 103:244-263.
- Manger WM, Gifford RW, Hoffman BB. Pheochromocytoma: a clinical and experimental overview. Curr Probl Cancer. 1985;9(5):1-89.
- Kantorovich V, Pacak K. Pheochromocytoma and paraganglioma. Prog Brain Res. 2010; 182:343-373. doi:10.1016/S0079-6123(10)82015-1
- Sarathi V. Characteristics of Pediatric Pheochromocytoma/paraganglioma. Indian J Endocrinol Metab. 2017;21(3):470–474. doi:10.4103/ijem.IJEM_558_16.
- Bholah R, Bunchman TE. Review of Pediatric Pheochromocytoma and Paraganglioma. Front Pediatr. 2017;5. doi:10.3389/fped.2017.00155
- Därr R, Lenders JWM, Hofbauer LC, Naumann B, Bornstein SR, Eisenhofer G. Pheochromocytoma – update on disease management. Ther Adv Endocrinol Metab. 2012;3(1):11-26. doi:10.1177/2042018812437356
- Lenders JWM, Duh Q-Y, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. doi:10.1210/jc.2014-1498
- Čtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15(4):3151-3160. doi:10.3892/etm.2018.5871
- Tsushima Y, Takahashi-Taketomi A, Endo K. Diagnostic utility of diffusion-weighted MR imaging and apparent diffusion coefficient value for the diagnosis of adrenal tumors. J Magn Reson Imaging. 2009;29(1):112-117. doi:10.1002/jmri.21616
- Alface MM, Moniz P, Jesus S, Fonseca C. Pheochromocytoma: clinical review based on a rare case in adolescence. BMJ Case Rep. 2015;2015. doi:10.1136/bcr-2015-211184
- Zelinka T, Musil Z, Dušková J, et al. Metastatic pheochromocytoma: clinical, genetic, and histopathologic characteristics. Eur J Clin Invest. 2011;41(10):1121-1128. doi:10.1111/j.1365-2362.2011.02518.x
- Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23(34):8812-8818. doi:10.1200/JCO.2005.03.1484
- Erlic Z, Neumann HPH. Familial pheochromocytoma. Hormones (Athens). 2009;8(1):29-38. doi:10.14310/horm.2002.1219
- Pham TH, Moir C, Thompson GB, et al. Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics. 2006;118(3):1109-1117. doi:10.1542/peds.2005-2299
- Bausch B, Wellner U, Bausch D, et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr Relat Cancer. 2014;21(1):17-25. doi:10.1530/ERC-13-0415.
Affiliations: Ohio University Heritage College of Osteopathic Medicine (Dr Saeed); Phoenix Children’s Hospital (Dr R Towbin); Cincinnati Children’s Hospital and University of Cincinnati College of Medicine (Dr A Towbin).