Middle interhemispheric variant of holoprosencephaly
By Sharma R, O’Brien WT, Guerin J, Kline-Fath BM, Towbin RB, Towbin AJ
CASE SUMMARY
A preterm baby girl born at 33 weeks’ gestation to a mother with maternal diabetes and a history of perinatal cocaine-use was diagnosed prenatally with midline interhemispheric variant (MIHV) of holoprosencephaly via ultrasonography and fetal MRI at 19 weeks’ gestation. At birth, the patient exhibited a prominent forehead, hypotelorism, a short upturned nose, and short palpebral fissures. While she exhibited difficulty feeding, the remainder of her neurological examination was normal.
IMAGING FINDINGS
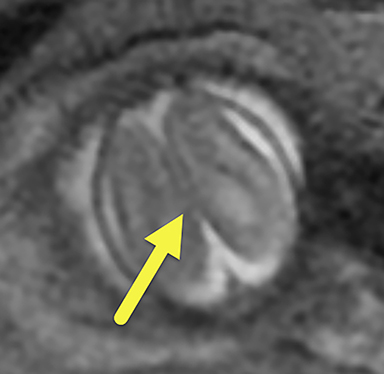
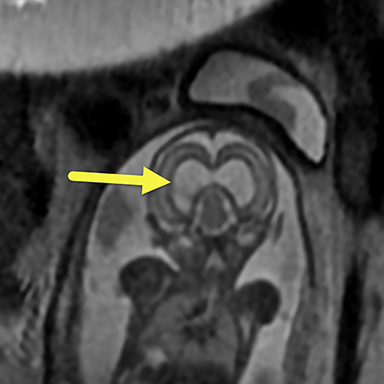
On fetal MRI (Figure 1), there was focal enlargement of the posterior horns of the lateral ventricles with the left measuring 12 mm and right 11mm. There was absence of the cavum vergae with continuity of the lateral ventricles across midline. The falx cerebri was partially absent in the posterior frontal/anterior parietal lobes with fusion of brain parenchyma across midline. A portion of the mid body of the corpus callosum was absent and the hippocampi were abnormally rotated.
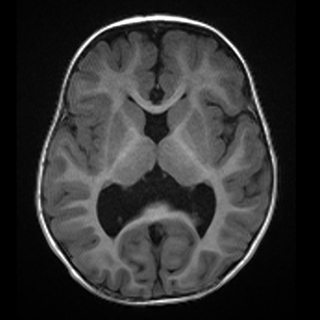
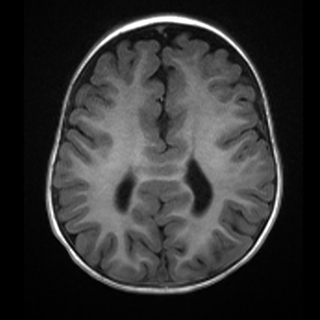
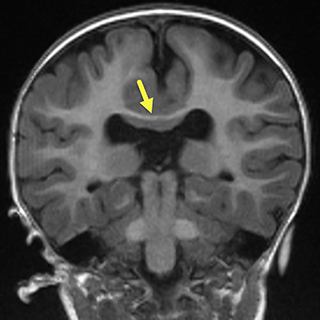
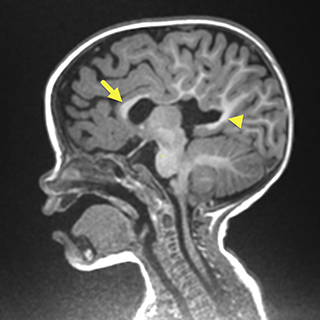
On postnatal brain MRI (Figure 2), the septum pellucidum and majority of the corpus collosum body and rostrum were absent resulting in continuity of the lateral ventricles across the midline and colpocephaly.
The falx cerebri was again seen to be partially absent in the area of the posterior frontal and anterior parietal lobes with associated continuity of the cortex across midline in this region. Associated anomalies visible on MRI included frontal lobe cortical malformations with thickened gyri and shallow sulci, as well as periventricular nodular gray matter heterotopia.
The thalami were closely approximated but not fused. The fornices were absent and the hippocampi were abnormally rotated. Evaluation of the sella revealed an ectopic neurohypophysis at the superior infundibulum.
DIAGNOSIS
Middle interhemispheric (AKA syntelencephaly) variant of holoprosencephaly
DISCUSSION
Holoprosencephaly (HPE) sequence is caused by failure of complete forebrain differentiation into two distinct cerebral hemispheres during gastrulation. Median facial structures are often involved and patients may exhibit seizures, hydrocephalus, motor and language dysfunction, endocrine abnormalities and respiratory issues which can occur as a result of aspiration due to difficulty swallowing.1,2 HPE has been associated with multiple genetic alterations, most notably trisomy 13, as well as other prenatal factors such as maternal diabetes (confers a 200-fold increase in risk), alcohol use, and CMV infection.
In order of increasing severity, the three main subtypes of HPE are lobar, semilobar, and alobar, all of which have varying degrees of separation of the cerebral hemispheres.2,3 Severity is based on several factors, which include the degree forebrain malformation, craniofacial malformation, seizure activity, hydrocephalus, and speech and ambulatory capabilities.4
Lobar HPE is the least severe subtype, characterized by noncleavage of the inferior aspect of the frontal lobes. The midline interhemispheric fissure, falx and posterior body and splenium of the corpus callosum are present and the thalami are usually separated. All types of HPE have absence of the septum pellucidum; if this is the only imaging finding on early prenatal imaging, this subtype can be difficult to recognize.5 Clinically, many patients require assistance with ambulation, have mild loss of hand function1 and are unable to speak full sentences. Craniofacial malformations are often subtle, and include microcephaly, hypotelorism, and a single maxillary central incisor.4
In the semilobar variant, the thalami, hypothalamus and at least half of the frontal lobes remain unseparated with resulting absence of the interhemispheric fissure and falx cerebri anteriorly.5 As in all types of HPE, the septum pellucidum is absent. The semilobar subtype of HPE also has mild associated facial abnormalities including hypotelorism and cleft lip. Patients with this subtype often lack the ability to ambulate effectively.4 They may develop rudimentary receptive language skills though production of normal speech is typically impaired.5
The alobar HPE variant is the most severe form of HPE. There is noncleavage of the cerebral hemispheres, thalami, hypothalamus and basal ganglia; this results in a single primitive monoventricle which communicates with a large dorsal cyst. Brain parenchyma may have a “ball” (most common), “cup” or “pancake” configuration, depending on severity.5 Craniofacial anomalies are more severe and include synopthalmia and a proboscis. Due to these more severe anomalies, patients with alobar HPE rarely survive the postnatal period.4
The middle interhemispheric variant (MIHV) of holoprosencephaly is a rare manifestation of abnormal forebrain differentiation in which there is impaired cleavage of the posterior frontal and anterior parietal lobes over the vertex.6-10 Anterior and posterior to this connection, the cerebral hemispheres are separated.7 Contrary to classic forms of HPE, MIHV represents a partial/focal lack of dorsal-ventral induction.8,9 The body of the corpus callosum is typically absent in the area of noncleavage, the Sylvian fissures are often vertically oriented and connected across midline and two-thirds have cortical dysplasia or subcortical heterotopic gray matter.5,6 In a study of 21 patients with MIHV, 86% of patients had abnormally oriented and connected Sylvian fissures and heterotopic gray matter. In all 21 subjects, the midline third ventricle separated the hypothalamus and lentiform nuclei.6
Survival of MIHV depends on several factors including the presence of facial and brain malformations, chromosomal abnormalities, and the involvement of other organs. Developmentally, patients with MIHV of HPE have similar disabilities compared to patients with the lobar HPE variant. Spacticity is the most common motor abnormality8 and patients are able to maintain speech and function with only mild impairment.4
CONCLUSION
The MIHV of HPE is a rare variant of HPE. It can be diagnosed via prenatal ultrasonography and MRI. Several features, such as the presence of a well formed anterior interhemispheric fissure, midline fusion of the posterior frontal and anterior parietal lobes with relative sparing of the basal forebrain, and pattern of mid-callosal partial agenesis and dysgenesis help distinguish this variant of HPE from the other subtypes on imaging. Developmentally, patients with the MIHV maintain speech function and are able to ambulate with assistance. Craniofacial malformation may be present but are mild in comparison to the more severe forms of HPE.
REFERENCES
- Levey, E.B., et al. Management of children with holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010; 154C(1): 183-190.
- Demyer, W., W. Zeman, and C.G. Palmer. The Face Predicts the Brain: Diagnostic Significance of Median Facial Anomalies for Holoprosencephaly (Arhinencephaly). Pediatrics. 1964. 34: 256-263.
- Hahn, J.S. and P.D. Barnes. Neuroimaging advances in holoprosencephaly: Refining the spectrum of the midline malformation. Am J Med Genet C Semin Med Genet. 2010; 154C(1): 120-132.
- Raam, M.S., B.D. Solomon, and M. Muenke. Holoprosencephaly: a guide to diagnosis and clinical management. Indian Pediatr. 2011; 48(6): 457-466.
- Winter, T.C., A.M. Kennedy, and P.J. Woodward, Holoprosencephaly: a survey of the entity, with embryology and fetal imaging. Radiographics. 2015. 35(1): 275-290.
- Simon, E.M., et al., The middle interhemispheric variant of holoprosencephaly. AJNR Am J Neuroradiol. 2002; 23(1): 151-156.
- Barkovich, A.J. and D.J. Quint. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. AJNR Am J Neuroradiol. 1993; 14(2): 431-440.
- Lewis, A.J., et al. Middle interhemispheric variant of holoprosencephaly: a distinct cliniconeuroradiologic subtype. Neurology. 2002. 59(12): 1860-1865.
- Bulakbasi, N., O. Cancuri, and M. Kocaoglu. The middle interhemispheric variant of holoprosencephaly: magnetic resonance and diffusion tensor imaging findings. Br J Radiol. 2016; 89(1063): 20160115.
- Picone, O., et al. Prenatal diagnosis of a possible new middle interhemispheric variant of holoprosencephaly using sonographic and magnetic resonance imaging. Ultrasound Obstet Gynecol. 2006; 28(2): 229-231.
Prepared by Dr. Sharma while at the University of Cincinnati College of Medicine, Department of Radiology; Dr. O’Brien, Dr. Guerin, Dr. Kline-Fath, and Dr. Alexander Towbin while at Cincinnati Children’s Hospital and the University of Cincinnati College of Medicine, Cincinnati, OH; and Dr. Richard Towbin while at Phoenix Children’s Hospital, Phoenix, AZ.