Multifocal amyloidoma
By Cassuto J, Siegel Y, Villada G, Villamizar N

CASE SUMMARY
A 59-year-old African American woman with a medical history of hypertension and gastroesophageal reflux disease presented to the emergency department with progressively worsening dyspnea, chest pain, epigastric pain, and orthopnea. Vital signs were temperature 36.9 C; respiratory rate 18 breaths/min, heart rate 99 bpm; blood pressure 127/85 mmHg; and SpO2 99% on room air. The patient did not demonstrate signs of distress during physical exam. Auscultation of the lungs demonstrated left sided crackles to the mid chest. Cardiovascular and abdominal exams were unremarkable. There was no peripheral edema. CBC and Chem7 values were within normal limits.
IMAGING FINDINGS
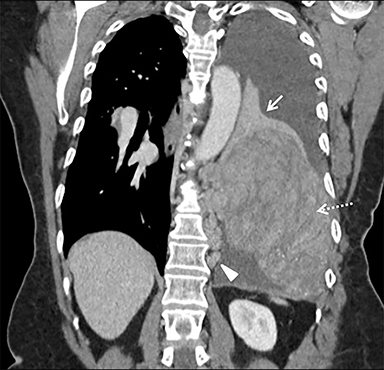
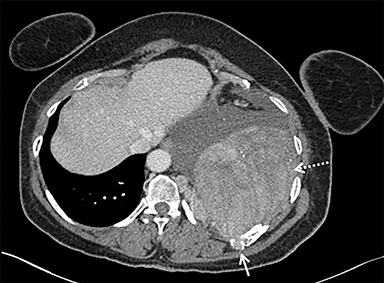
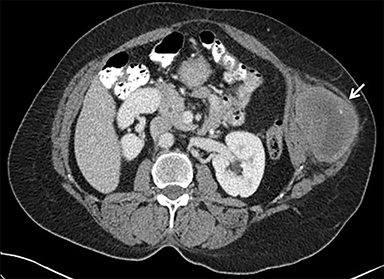
A chest radiograph demonstrated a large left pleural effusion with minimal aeration of the left upper lobe. There was a dense opacity at the left base. Chest, abdomen, and pelvis CT (Figure 1) demonstrated a left lower-thorax enhancing mixed-density mass with punctate calcifications measuring 13.3 × 10.0 × 15.9 cm. The mass involved the left 10th posterior rib resulting in a pathologic fracture. Multiple adjacent extra-pleural paraspinal smaller masses with similar CT characteristics to the large mass were also seen. A large left pleural effusion was also noted. Intra-abdominal findings were notable for a heterogeneous mass within the left abdominal wall, measuring 8.7 × 6.4 × 10.4 cm.
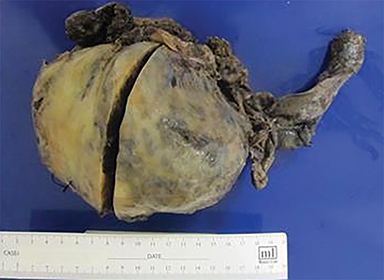
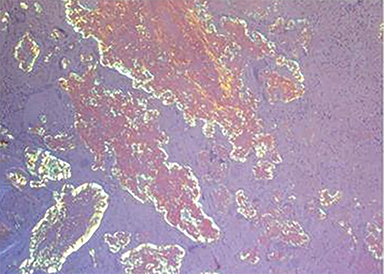
Percutaneous biopsy of the dominant intrathoracic and abdominal wall masses demonstrated congo red staining with apple-green birefringence under polarized light, large amyloid deposits with focal calcifications, and evidence of fibrosis – findings consistent with amyloidoma. The masses were surgically excised with the following findings: The intrathoracic mass was firmly attached to the chest wall at the level of the 10th rib and the left lower lobe. An additional multilobulated mass involving the intrathoracic fascia, intercostal muscles, and sympathetic chain was removed. The abdominal tumor was located deep to the left external oblique muscles and was non-infiltrating. Pathology (Figure 2) diagnosed the intrathoracic and abdominal wall masses as amyloidomas, sub-type AL. There was no evidence of associated neoplasm.
DIAGNOSIS
Multifocal amyloidoma.
Differential Diagnosis includes sarcoma, lymphoma with secondary amyloid deposition.
DISCUSSION
Amyloidosis is a rare disease with an annual incidence of 6-10 cases per 100,000 people in Western Europe.1 It is characterized by the pathologic deposition and accumulation of protein (amyloid) within organs or tissues. The disease may be characterized based on the site of deposition (systemic vs localized to one organ) or protein type (amyloid light chain vs amyloid A).2,3 Amyloid light chain (AL amyloidosis) results from the production and deposition of monoclonal immunoglobulin light chains by abnormal plasma cells. Reactive amyloid A (AA amyloidosis) deposition is a consequence of IL-6 mediated macrophage cleavage of serum amyloid A (produced by the liver) in the setting of chronic disease states.2,3
In brief, amyloid protein deposits are composed of abnormally folded serum amyloid P, glycosaminoglycans, and fibril proteins which fold into characteristic beta-pleated sheets. These abnormal protein deposits are insoluble, resistant to macrophage mediated degradation, and result in organ failure due to anatomic disruption of normal tissue function and organ blood supply. Alternatively, mass effect from the even rarer focal form (amyloidoma) may compromise organ function.1 Radiographically, there are many presentations of amyloidosis, such as diffuse or nodular intestinal wall thickening, organomegaly, left ventricular cardiac wall thickening, recurrent pneumonia in the same location, tree-in-bud and nodular pulmonary pattern, diffuse discontinuous bronchial wall calcifications, muscular hypertrophy (shoulder girdle is most common), synovial thickening, and focal mass mimicking neoplasm.1
Amyloidoma, the rarest form of amyloid deposition, is a discrete, focal, and tumor-like aggregate of amyloid deposition.4 The respiratory system, gastrointestinal tract, and central nervous system are often reported as the most common involved sites for amyloidomas. Case reports detailing amyloidomas of the soft tissues of the extremities, retroperitoneum, and mesentery have also been described.5-11 Amyloidosis presenting as a focal mass-like deposition has several radiologic phenotypes that have been described. These include: homogeneous soft-tissue density lesions, heterogeneous density with or without central necrosis, with micro or macrocalcifications, and as an infiltrating process.1,12,13 Several of these characteristics can be seen in aggressive diseases such as sarcoma, lymphoma, and metastasis.
Classic descriptions of amyloidomas detail solitary or multiple amyloid nodules within a single discrete anatomic site. Herein, we report a unique case of multifocal amyloidoma, involving both the abdominal wall and the thorax in separate discontinuous locations. In this case, the patient presented with a locally invasive intrathoracic chest-wall mass associated with a cluster of intrathoracic paraspinal smaller masses, and an abdominal soft-tissue mass. Tumor size and multifocal presentation were concerning for a neoplasm such as sarcoma or lymphoma with secondary amyloid deposition. Nodular amyloidoma of the lungs has been previously described associated with primary low grade lymphoma.14 Furthermore, lymphoma can present as chest or abdominal wall masses.15,16
CONCLUSION
Although this report is of a rare presentation of a rare disease, it is important, in the appropriate clinical setting, to add this entity to the gamut of multifocal masses involving mesenchymal tissue. While amyloidoma is a known entity typically confined to singular organ beds and compartments, to the best of our knowledge, this is the first reported case of a multifocal presentation.
REFERENCES
- Czeyda-Pommersheim F, Hwang M, Chen S. Amyloidosis: Modern cross-sectional imaging. Radiographics. 2014;35:1381-1392.
- Wechalekar AD, Gillmore JD, Hawkins. Systemic Amyloidosis. Lancet. 2016;387:2641-2654.
- Real de Asua D, Costa R, Galvan J, et al. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clin Epidemiol. 2014;6:369-377.
- Krishnan J, Chu WS, Elrod JP, et al. Tumoral presentation of amyloidosis (amyloidomas) in soft tissues. A report of 14 cases. Am J Clin Patho. 1993;100:135-144.
- Maheshwari AV, Muro-Cacho CA, Kransdorf M, et al. Soft-tissue amyloidoma of the extremities: a case report and review of literature. Skeletal Radiology. 2009;38:287-292.
- Werner BC, Shen FH, Shimer AL. Primary cervical amyloidoma: a case report and review of literature. Spine J. 2013;13:e1-7.
- Yokota K, Kishida D, Kayano H, et al. A case of abdominal aortic retroperitoneal and mesenteric amyloid light chain amyloidoma. Case Rep Rheumatol Epub. 2016:1-5.
- Kartal D, Canoz O, Balkanli, et al. Cutaneous amyloidoma: A rare case report. Ann Dermatol. 2016;28:643-645.
- Desai RA, Mahajan VK, Benjamin S, et al. Pulmonary amyloidoma and hilar adenopathy. Rare manifestations of primary amyloidosis. Chest. 1979;76:170-173.
- Gerami S, Easley GW, Payan H. Primary localized amyloidosis of the urethra and bladder (amyloidoma). Am Surg. 1970;36:375-377.
- Chin-Lenn L, Ryan C, Skene A et al. Proximal esophageal amyloidoma presenting with life threatening dysphagia. Ear Nose Throat J. 2009;88:E4.
- Parmar H, Rath T, Castillo M, et al. Imaging of focal amyloid depositions in the head, neck, and spine: Amyloidoma. Am J Neuroradiol. 2010;31:1165-1170.
- Prause CA, Zhai Q, Weindling SM. Pharyngeal amyloidomas: Variable appearance on imaging. Neuroradiol. 2017;30:235-239.
- Dacic S, Colby T, Yousem S. Nodular amyloidoma and primary pulmonary lymphoma with amyloid production: A differential diagnostic problem. Mod Pathol. 2000;13:934-940.
- Witte B, Hurtgen M. Lymphomas presenting as chest wall tumors. Thorac Surg Sci. 2006;3:1-4.
- Bozas G, Anagnostou D, Tassidou, et al. Extranodal non-Hodkin’s lymphoma presenting as an abdominal wall mass. A case report and review of literature. Leuk Lymphoma. 2006;47:329-332.
Prepared by Dr. Cassuto and Dr. Siegel while practicing in the Department of Radiology; Dr. Villada while practicing in the Department of Pathology; and Dr. Villamizar while practicing in the Division of Cardiothoracic Surgery, all at Jackson Memorial Hospital, Miller School of Medicine, University of Miami, Miami, FL.