Wernicke Encephalopathy
Images


CASE SUMMARY
A 53-year-old with a history of recently diagnosed metastatic pancreatic cancer was admitted with complaints of worsening diplopia, ataxia, and altered mental status. Following the diagnosis, the patient acknowledged drinking heavily owing to abdominal pain and depression.
IMAGING FINDINGS
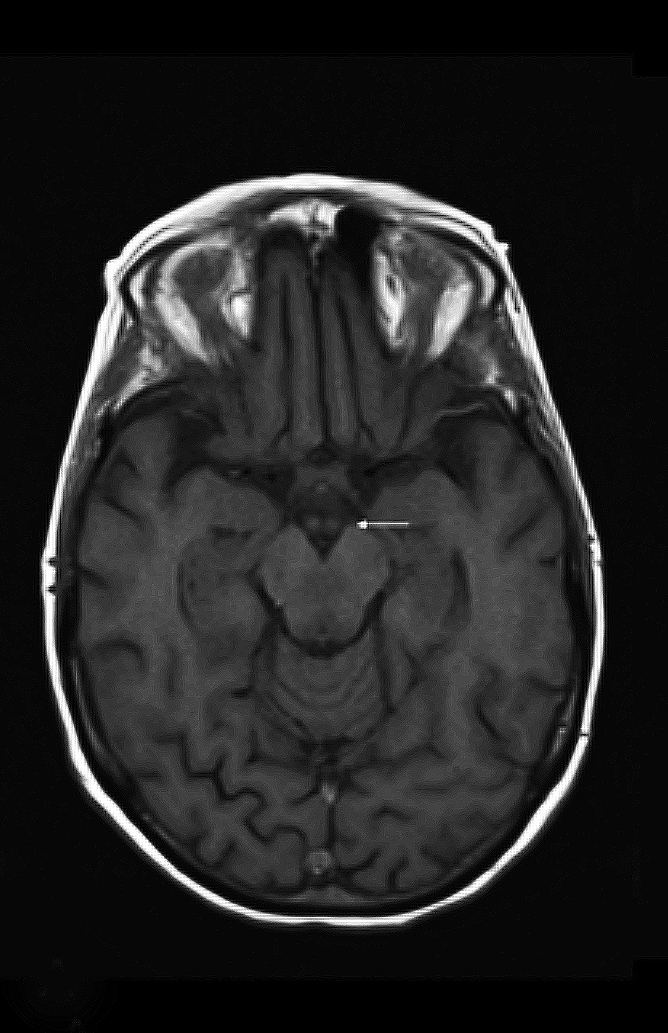
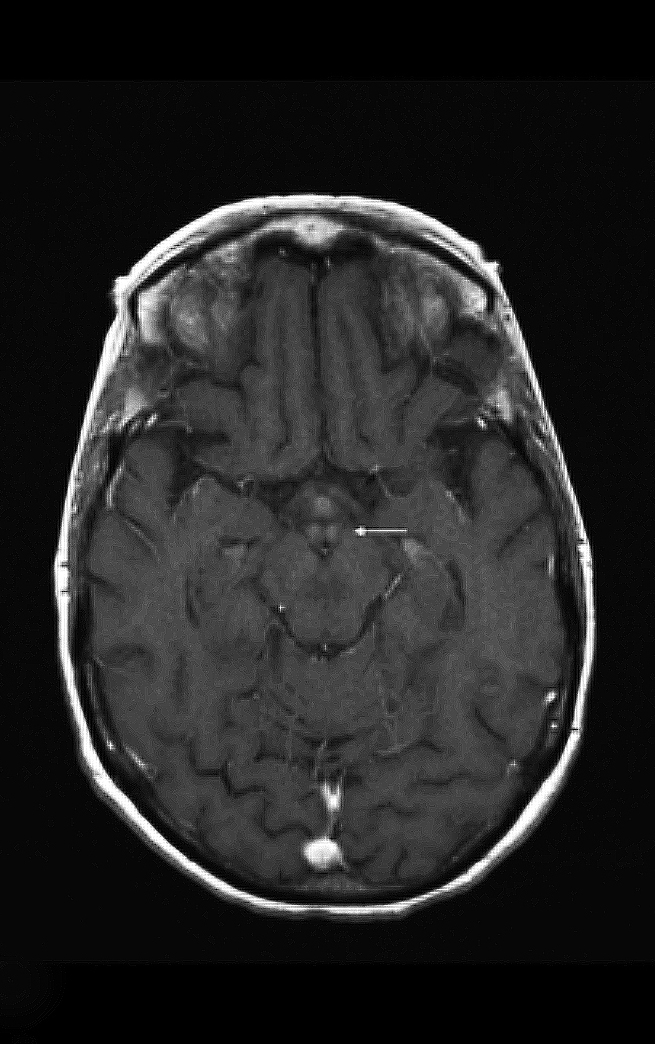
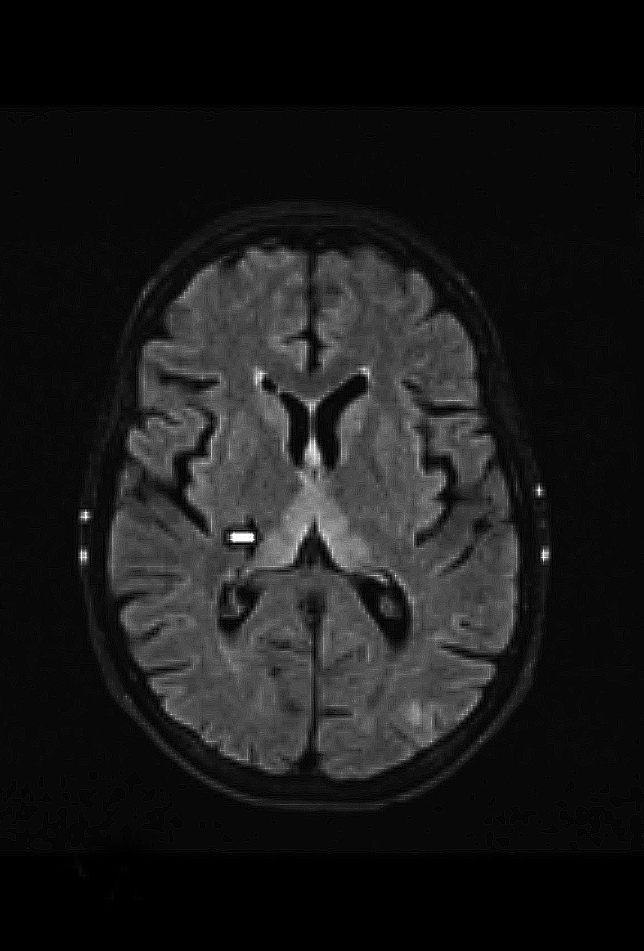
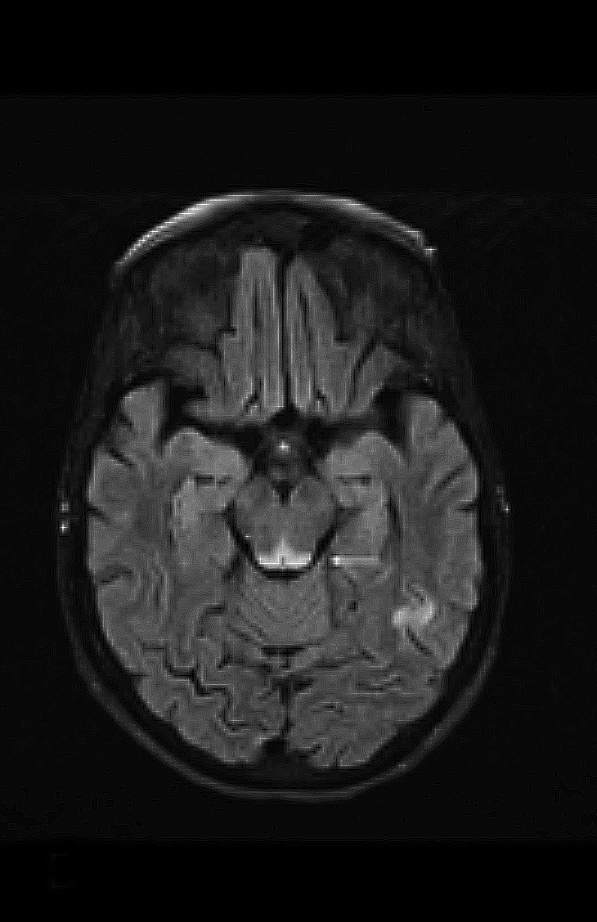
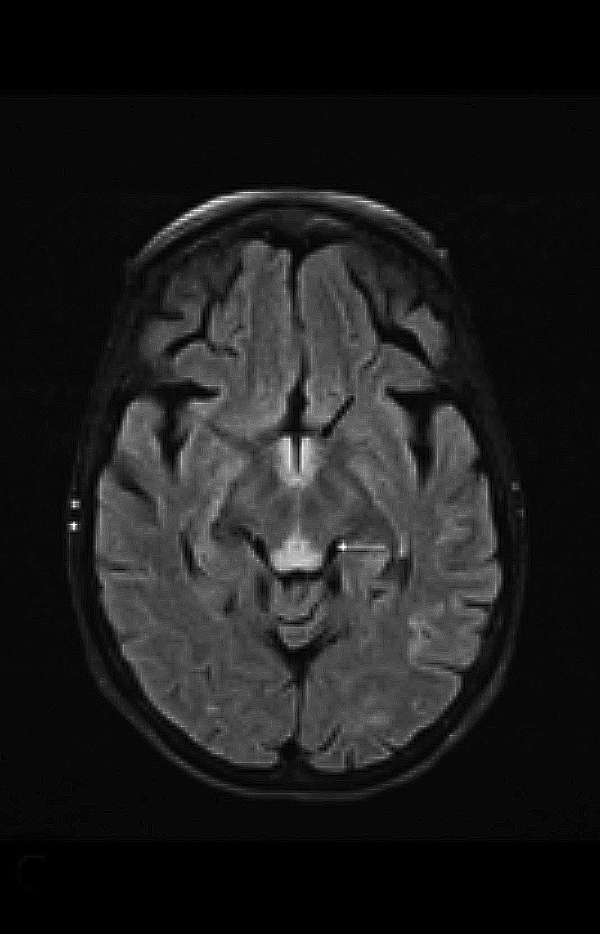
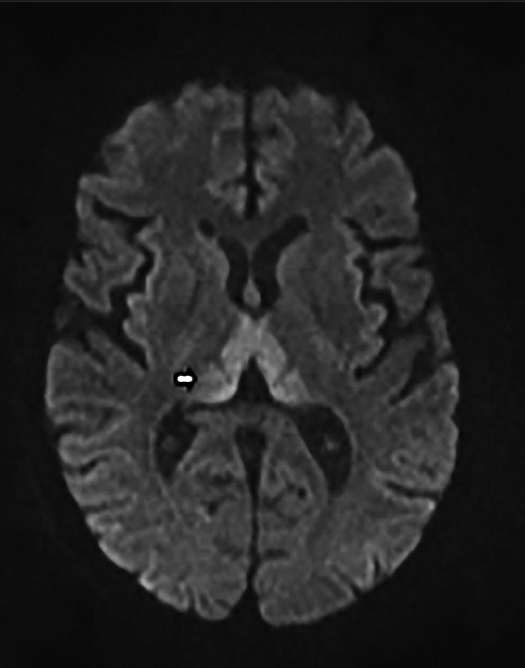
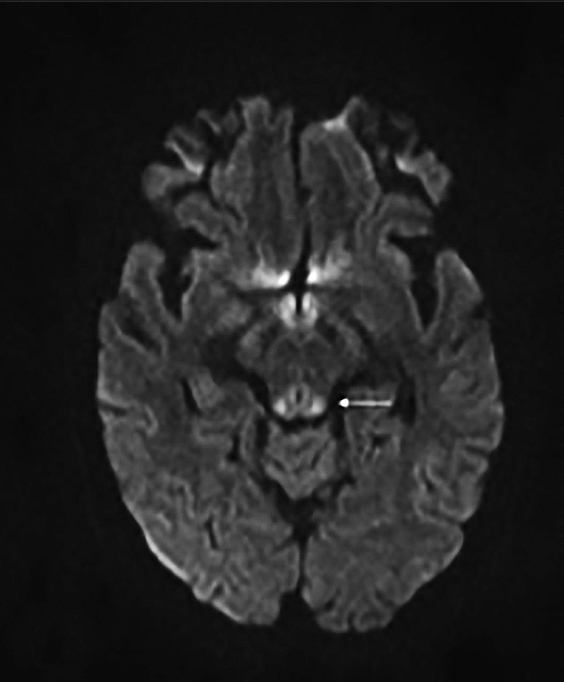
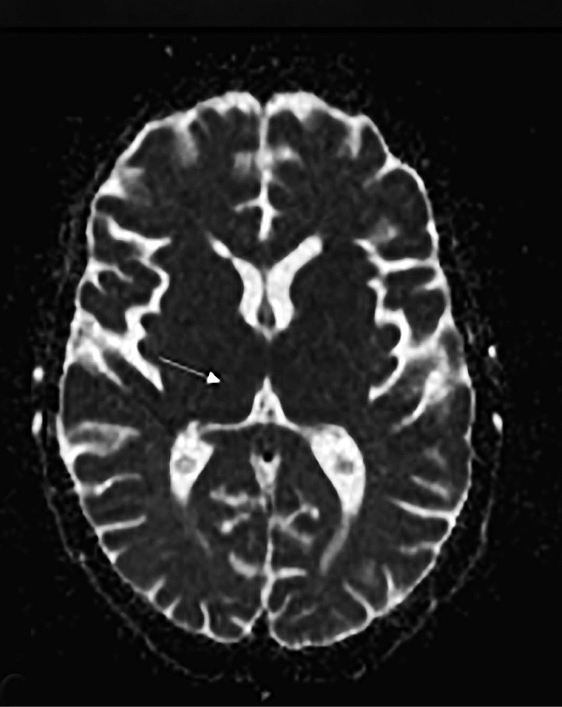
Upon admission, pre- and postcontrast brain MRI scans demonstrated symmetric bilateral increased T2 signal in the medial thalami, hypothalamus, periaqueductal (PA) white matter in the left occipital and temporal lobes. Postcontrast imaging demonstrated faint enhancement in the bilateral mammillary bodies. Diffusion weighted imaging also revealed restricted diffusion in the bilateral medial thalami and periaqueductal gray matter.
DIAGNOSIS
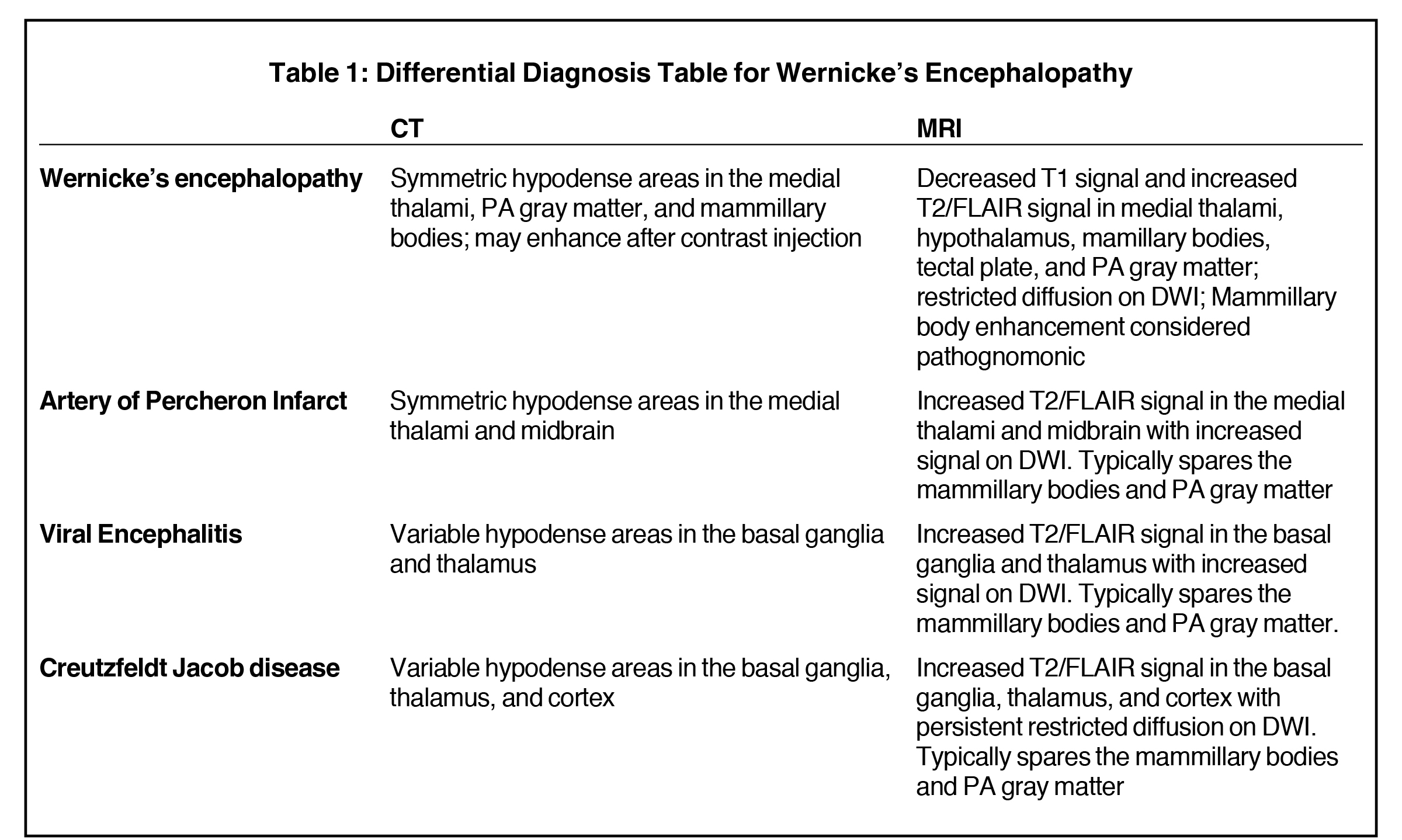
Wernicke encephalopathy. Differential diagnoses: Artery of Percheron (AoP) infarct, viral encephalitis, and Creutzfeldt-Jacob disease (CJD).
DISCUSSION
Wernicke encephalopathy (WE) was first described in 1881 by Carl Wernicke, and is defined as a neurologic disorder attributable to thiamine deficiency most often related to chronic alcohol consumption.5 The condition is fairly common, with some studies reporting an incidence of 2.8% in the general population and a prevalence as high as 12.5% in alcohol abusers.6 It most often presents as the classic triad of encephalopathy, oculomotor dysfunction, and gait ataxia.
Imaging is useful in cases of suspected WE, as abnormal findings may help rule out competing diagnoses. Head CT may depict lesions as symmetrical hypodense areas in the medial thalami, PA gray matter, and mammillary bodies that may enhance after contrast injection.
MRI, however, is the most sensitive imaging modality for WE. In the acute setting, WE on MRI typically demonstrates decreased T1 signal (Figure 1) and increased T2 (Figure 2) signal in these regions, as well as restricted diffusion on diffusion weighted imaging (Figure 3). Most of these signal abnormalities are located in the medial thalami, hypothalamus, mammillary bodies, tectal plate, and the periventricular white matter.1
Mammillary body enhancement is common in acute WE and considered pathognomonic. Indeed, mammillary body atrophy is a relatively specific indicator of chronic WE. Charness, et al, demonstrated that 78% of patients with WE had smaller mammillary bodies than matched controls and patients with Alzheimer dementia.3
Petechial hemorrhage in the medial thalami and mammillary bodies may be observed in severe cases of acute WE. In chronic WE, the mammillary bodies and cerebellar vermis tend to be atrophic without the T2 hyperintense signal intensity observed.2
An AoP infarct demonstrates symmetric hypodense areas in the medial thalami and midbrain on CT and corresponding areas of increased T2 signal and restricted diffusion on DWI on brain MRI. However, an AoP infarct typically spares the mammillary bodies and periaqueductal gray matter, unlike acute WE. Numerous viral encephalitides, including West Nile, Japanese encephalitis, and St. Louis encephalitis, may variably involve the thalami and basal ganglia producing hyperintense T2 signal in those locations with restricted diffusion on DWI, but otherwise spares the mammillary bodies and periaqueductal gray matter. Finally, CJD, the spongioform encephalopathy, causes increased T2 signal in the basal ganglia, thalami, and cortex with persistent restricted diffusion on DWI but will also spare the mammillary bodies and periaqueductal gray matter (Table 1). While brain MRI may aid in the diagnosis of WE and exclude competing diagnoses, this disease is predominantly a clinical diagnosis. The use of the Caine criteria dietary deficiency, oculomotor abnormalities, cerebellar dysfunction, and either altered mental status or mild memory impairment) has been shown to have a sensitivity of 85% when 2 out of the 4 symptoms are present.4
Adults are primarily affected with an even distribution between 30-70 years old. Fifty percent of cases are alcohol-related with 50% of cases attributable to non-alcoholic-related malabsorption, including bariatric surgery, thiamine-restrictive diets, starvation, hyperemesis gravidarum, gastric malignancy, and inflammatory bowel disease.
Long term alcohol consumption depletes thiamine stores by decreasing nutritional intake and causing malabsorption. Thiamine helps maintain the integrity of cellular membranes and is an important cofactor for several enzymes chiefly transketolase, alpha-ketoglutarate dehydrogenase, and pyruvate dehydrogenase. These enzymes participate in carbohydrate metabolism and dysfunction results in decreased cerebral energy utilization leading to neuronal injury (both intra- and extracellular edema) in parts of the brain with high metabolic demand.7 The lesions are characteristically bilateral and most commonly involve the mammillary bodies, dorsomedial thalami, locus ceruleus, periaqueductal gray matter, vestibular nuclei, and oculomotor nuclei.8
If WE is clinically suspected, treatment should begin promptly as progression to coma and death is common. Treatment involves high dose thiamine 500 mg infused over 30 minutes, three times daily for two consecutive days and 250 mg intravenously or intramuscularly once daily for an additional five days.9 Thiamine must be replaced before the administration of glucose as this may worsen the encephalopathy. MRI is helpful in the diagnosis and characterizing the chronicity of WE, while excluding other pathologies. When WE is suspected clinically however treatment should begin promptly, while failing to treat carries a high risk of progression to coma and death10 (Table 2).
CONCLUSION
MRI findings in the acute stage of WE include bilateral T2 hyperintensity in the mammillary bodies, medial thalami hypothalamus, and periaqueductal gray matter with variable restricted diffusion on DWI in the corresponding areas and enhancement of the mammillary bodies on postcontrast imaging.
REFERENCES
- Zuccoli G, Pipitone N. Neuroimaging Findings in Acute Wernicke’s Encephalopathy: Review of the Literature. American Journal of Roentgenology 2009 192:2, 501-508. PMID: 19155417.
- Park SH, Kim M, Na DL, Jeon BS. Magnetic resonance reflects the pathological evolution of Wernicke encephalopathy. J Neuroimaging. 2001;11(4):406. PMID: 11677881.
- Charness ME, DeLaPaz RL. Mamillary body atrophy in Wernicke’s encephalopathy: antemortem identification using magnetic resonance imaging. Ann Neurol. 1987;22(5):595. PMID: 3426166.
- Caine D, Halliday GM, Kril JJ, Harper CG. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62(1):51. PMID: 9010400
- Victor, M, Adams, RA, Collins, GH. The Wernicke-Korsakoff syndrome and related disorders due to alcoholism and malnutrition. FA Davis, Philadelphia 1989. ISBN 0803689217.
- Harper C, Fornes P, Duyckaerts C, Lecomte D, Hauw JJ. An international perspective on the prevalence of the Wernicke-Korsakoff syndrome. Metab Brain Dis. 1995 Mar;10(1):17-24. PMID: 7596325
- Martin PR, Singleton CK, Hiller-Sturmhöfel S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health. 2003;27(2):134. PMID: 15303623
- Torvik A. Two types of brain lesions in Wernicke’s encephalopathy. Neuropathol Appl Neurobiol. 1985;11(3):179. PMID: 3929155.
- Cook CC, Hallwood PM, Thomson AD. B Vitamin deficiency and neuropsychiatric syndromes in alcohol misuse. Alcohol Alcohol. 1998;33(4):317. PMID: 9719389.
- Galvin R, Bråthen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA, EFNS. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010 Dec;17(12):1408-18. PMID: 20642790.
Citation
P F, B E, P N, D R, S S, E S.Wernicke Encephalopathy. Appl Radiol. 2020; (3):48D-48G.
May 5, 2020