Synovial sarcoma
Images
CASE SUMMARY
A 26-year-old male presented with complaints of pain and swelling in the right lower thigh lasting a month. A mass lesion was detected in the posteromedial aspect of the lower thigh on magnetic resonance imaging (MRI). A biopsy followed by histopathological analysis confirmed the diagnosis suggested by the MRI. Neoadjuvant chemotherapy with doxorubicin and ifosfamide was administered. A follow-up MRI after 6 cycles of chemotherapy showed a remarkable reduction in the tumor size.
DIAGNOSIS
Synovial sarcoma
Differential diagnosis: Malignant fibrous histocytoma, fibrosarcoma, liposarcoma, osteosarcoma, and chondrosarcoma.
IMAGING FINDINGS
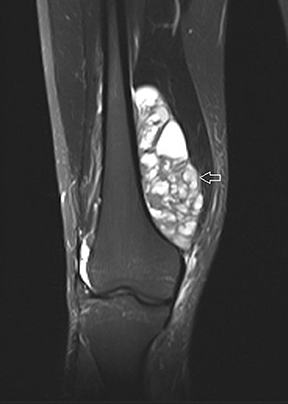
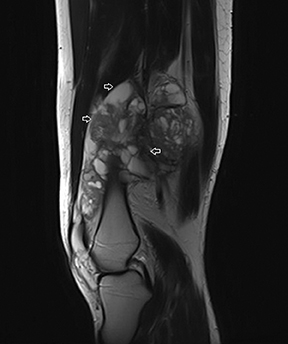
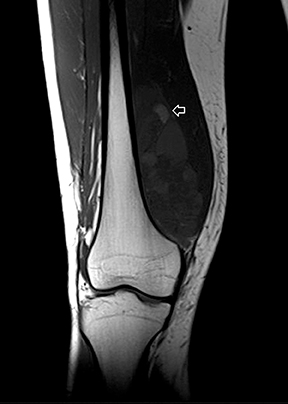
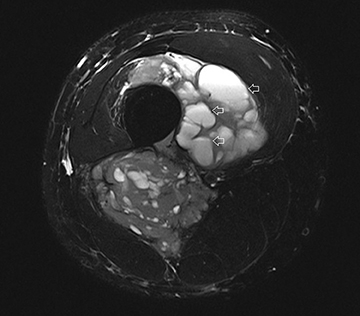
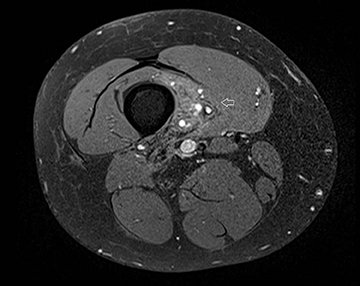
The coronal proton density fat-saturated (PDFS) MR sequence (Figure 1) reveals a large mass lesion in the postero-medial region of the right lower thigh, measuring about 15 × 10 × 7 cm. The mass shows multilocular cystic areas, which appear hyperintense on T2-weighted(T2W) (Figure 2) and PDFS (Figure 1) sequences, along with solid components that appear hypointense and isointense on T2W sequence(Figure 2). Some of the cystic areas reveal hyperintense signal on T1-weighted (T1W) sequence (Figure 3), suggestive of the presence of hemorrhagic foci. The axial PDFS sequence (Figure 4) reveals fluid-fluid levels in some of the cystic areas. The mass is located deep to the vastus medialis and hamstring muscles, abutting the distal femoral diaphysis without any signal abnormality in the bone, as seen on the axialPDFS sequence (Figure 4). The follow-up MRI after 6 cycles of chemotherapy showed remarkable reduction in tumor size, as seen on the axial PDFS sequence (Figure 5).
DISCUSSION
Synovial sarcoma is one of the most common soft-tissue tumors in adolescents and young adults. The tumor usually develops in the first 3 decades of life. Though the most common site of occurrence is around the knee joint, it can involve any part of appendicular skeleton. Other rarer sites of involvement include the head and neck region and the chest wall.
The origin of synovial sarcoma is unclear. The name ‘synovial sarcoma’ was coined because of the microscopic similarity of some tumors to synovium and their propensity to arise near joints. However, the actual cells from which the tumor develops are unknown and not necessarily synovial. A neurologic origin has been suggested.1 A histologic resemblance has been found between the neural cells of malignant peripheral nerve sheath tumor and synovial cell sarcoma.1 The pathophysiology involves a gene translocation abnormality t(X;18) (p11.2;q11.2).2
Synovial sarcoma is graded using the widely accepted grading scheme for all sarcomas, the FNCLCC (French Federation Nationale desCentres de Lutte Contre le Cancer) scheme, which uses a combined score from 3 separate parameters, including degree of differentiation,mitotic activity, and necrosis. Both biphasic and monophasic synovial sarcomas are usually intermediate grade (grade 2/3); however, both types can be high grade (grade 3/3).3 Poorly differentiated synovial sarcomas are high-grade tumors.
Though plain radiographs and CT may aid in the diagnosis, MRI is the imaging modality of choice for evaluating soft-tissue sarcomas. Staging requires local imaging with MRI, whereas the likely site of distant spread (the lungs) can be evaluated with CT. Though not very specific, a combination of the following signs in an appropriate clinical setting (young age, juxta-articular location) suggest a diagnosis of synovial sarcoma on MR imaging:4,5
Triple sign – Areas of high, intermediate, and low signal intensity on T2W images. The appearance is presumably the result of the mixture of solid cellular elements (intermediate signal intensity), hemorrhage or necrosis (high signal intensity), and calcified or fibrotic collagenized regions (low signal intensity). The triple sign has been described as occurring in 35% to 57% of cases in larger series.4.5 However,the triple sign is also seen in other soft-tissue neoplasms, particularly in malignant fibrous histiocytoma; and hence it is not specific for synovial sarcoma.
Bowl of grapes sign — The clustering of largely cystic areas creates a bowl of grapes appearance.
Fluid – fluid levels — Intra-tumoral hemorrhage is responsible for fluid levels. Fluid levels have been described in 10% to 25% of synovial sarcomas in several series.4
A large tumor size ( ≥ 5 cm) has been consistently proven to be associated with development of distant metastasis and decreased disease-specific survival.6 Other indicators of poor prognosis include tumor location in the trunk or head and neck, older patients, cystic / hemorrhagic components, marked heterogeneity, and histologic features, suggesting a high-grade tumor.6,7
Many studies have shown neoadjuvant chemotherapy (doxorubicin, ifosfamide, and mensa) to improve disease-specific survival,particularly in patients with extremity tumors of a large size (> 5 cm).6,7,8 Though neoadjuvant radiotherapy is proposed in some cases to reduce tumor size, it is associated with an increased rate of wound problems. Surgery is the mainstay of treatment. A tumor-free margin of 1 cm to 3 cm is recommended.9 MRI is extremely helpful in preoperative planning of tumor excision. Postoperative radiation therapy is usually required, particularly if the margins are close to vital neurovascular structures. The most common radiotherapy is external-beam radiation directed at the tumor site, including a margin of surrounding normal tissue.
CONCLUSION
In conclusion, synovial sarcoma is an aggressive tumor and one of the most common soft-tissue neoplasms, which occur in the first 3decades of life. It has a propensity to involve juxta-articular regions. A large tumor size at the time of diagnosis indicates poor prognosis. MRI is the imaging modality of choice in the preoperative evaluation of these lesions. Neoadjuvant chemotherapy has been found to improve disease-specific survival in some patient groups; particularly those with large-sized extremity tumors. Surgery is the mainstay of treatment.
REFERENCES
- Ladanyi M, Antonescu CR, Leung DH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: A multi-institutional retrospective study of 243 patients. Cancer Res. 2002;62:135-140.
- Kawai A, Woodruff J, Healey JH, et al. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998;338:153-160.
- Coindre JM, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: A study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91:1914-1926.
- Murphey MD, Gibson MS, Jennings BT, et al. From the archives of AFIP: Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26:1543-1565.
- O’Sullivan PJ, Harris AC, Munk PL. Radiological features of synovial cell sarcoma. Br J Radiol. 2008;81:346-356.
- Eilber FC, Brennan MF, Eilber FR, et al. Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann Surg. 2007;246:105-113.
- Grobmyer SR, Maki RG, Demetri GD, et al. Neo-adjuvant chemotherapy for primary high-grade extremity soft tissue sarcoma. Ann Oncol. 2004;15:1667-1672.
- Siehl J, Thiel E, Schmittel A, et al. Ifosfamide/liposomal daunorubicin is a well tolerated and active first-line chemotherapy regimen in advanced soft tissue sarcoma. Cancer. 2005;104:611-617.
- Zagard G, Ballo M, Pisters P, et al. Prognostic factors for patients with localized soft tissue sarcoma treated with conservation surgery and radiation therapy. Cancer. 2003;97:2530-2543.
Citation
C S. Synovial sarcoma. Appl Radiol. 2013;(11):42-44.
November 4, 2013