Sellar Atypical Teratoid Rhabdoid Tumor
Images
Case Summary
A young adult woman with no significant medical history presented with diplopia, right mydriasis, and right ophthalmalgia. The patient also endorsed a history of extreme migraine headache and right facial paresthesias. Symptoms were progressive over a
2-year period. Laboratory evaluation was unremarkable.
Imaging workup revealed a large, parasellar mass. The patient subsequently underwent staged surgical resection. Following the second staged procedure, the patient underwent repeat imaging, which demonstrated extensive areas of cerebral vasospasm. Two-month follow-up imaging demonstrated tumor progression.
Imaging Findings
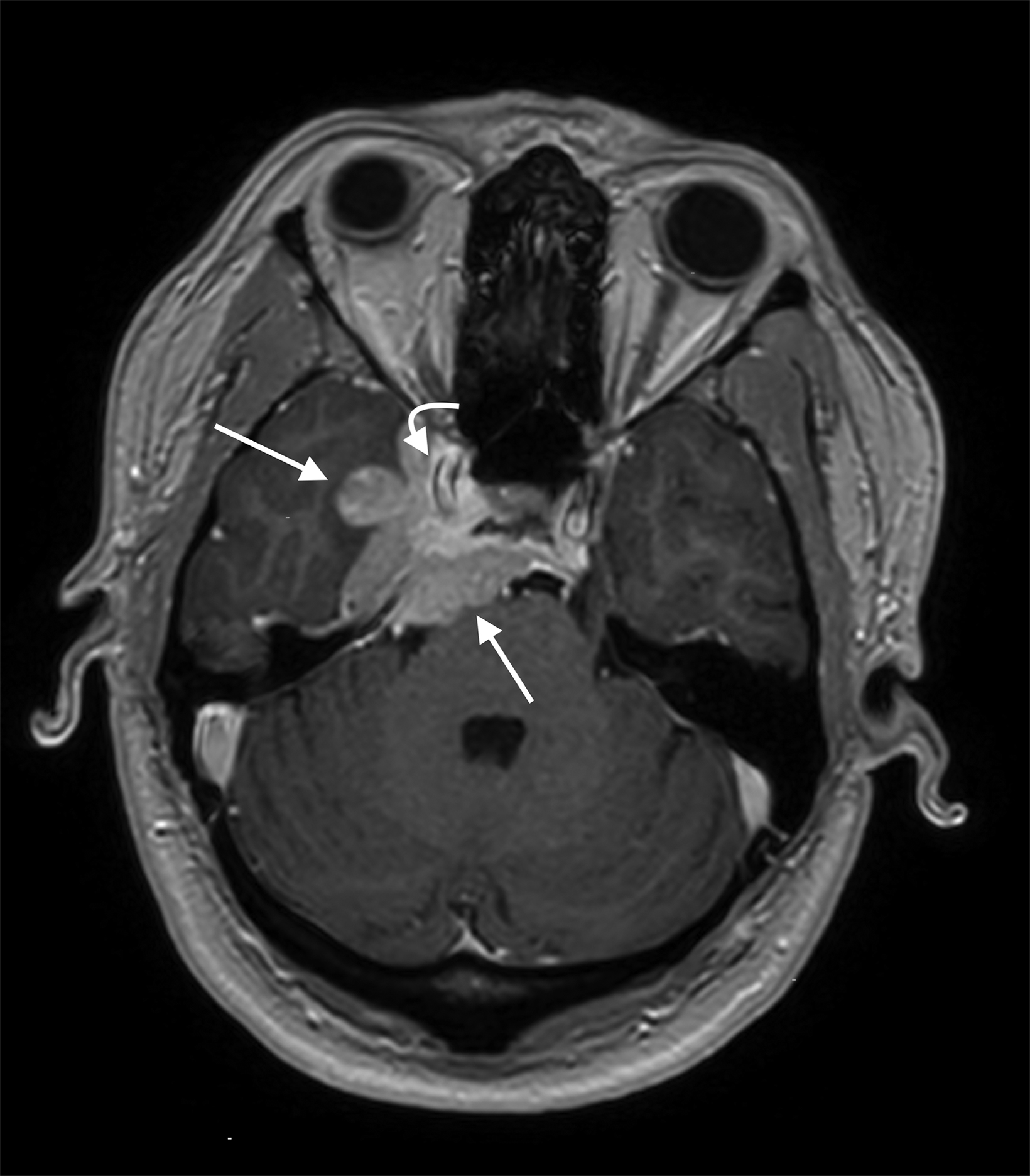
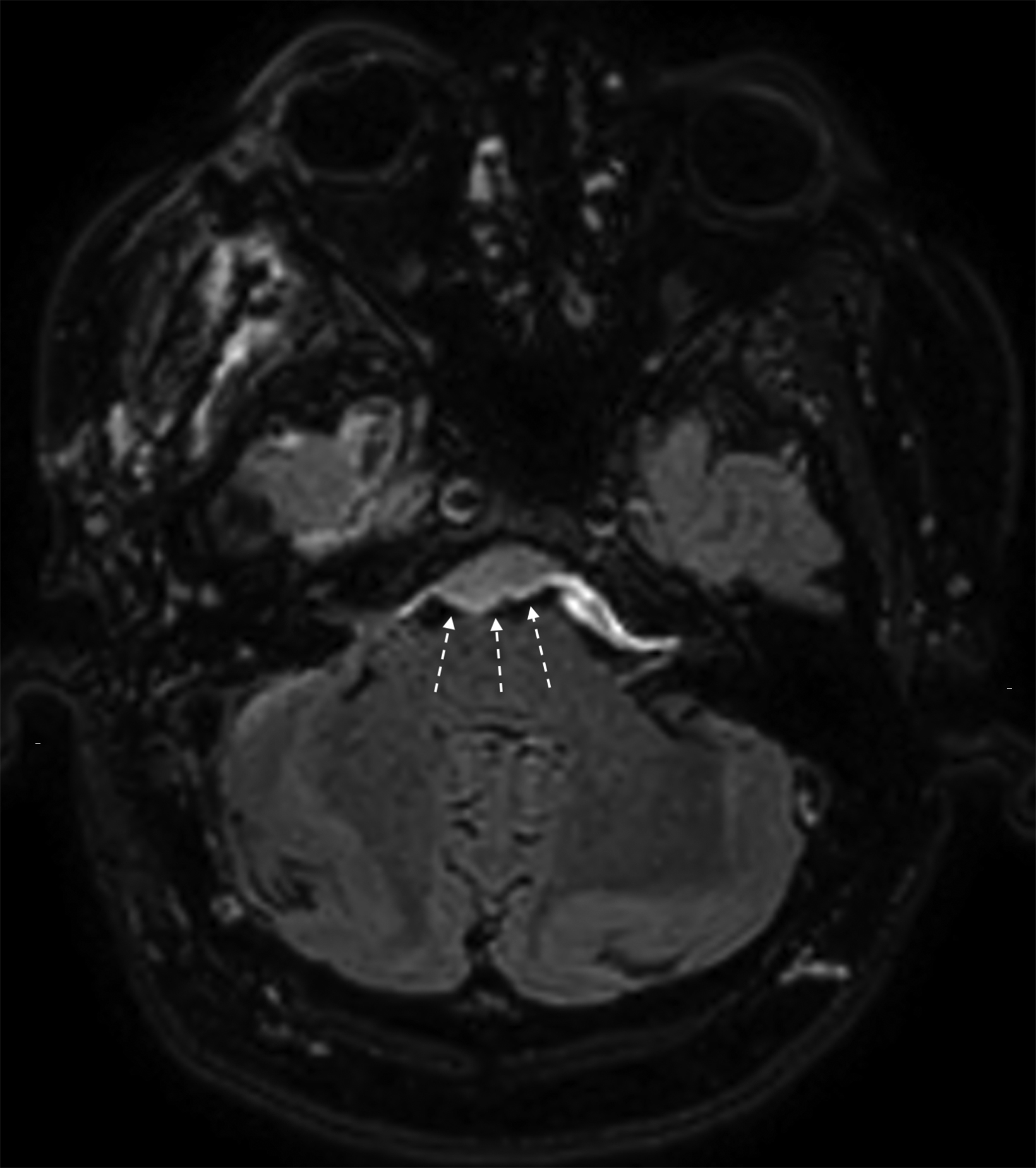
Preoperative MRI (Figure 1) demonstrated a large, contrast-enhancing, T2/FLAIR hyperintense, dural-based mass in the right parasellar region. The mass encased and narrowed the right internal carotid artery and abutted the basilar artery.
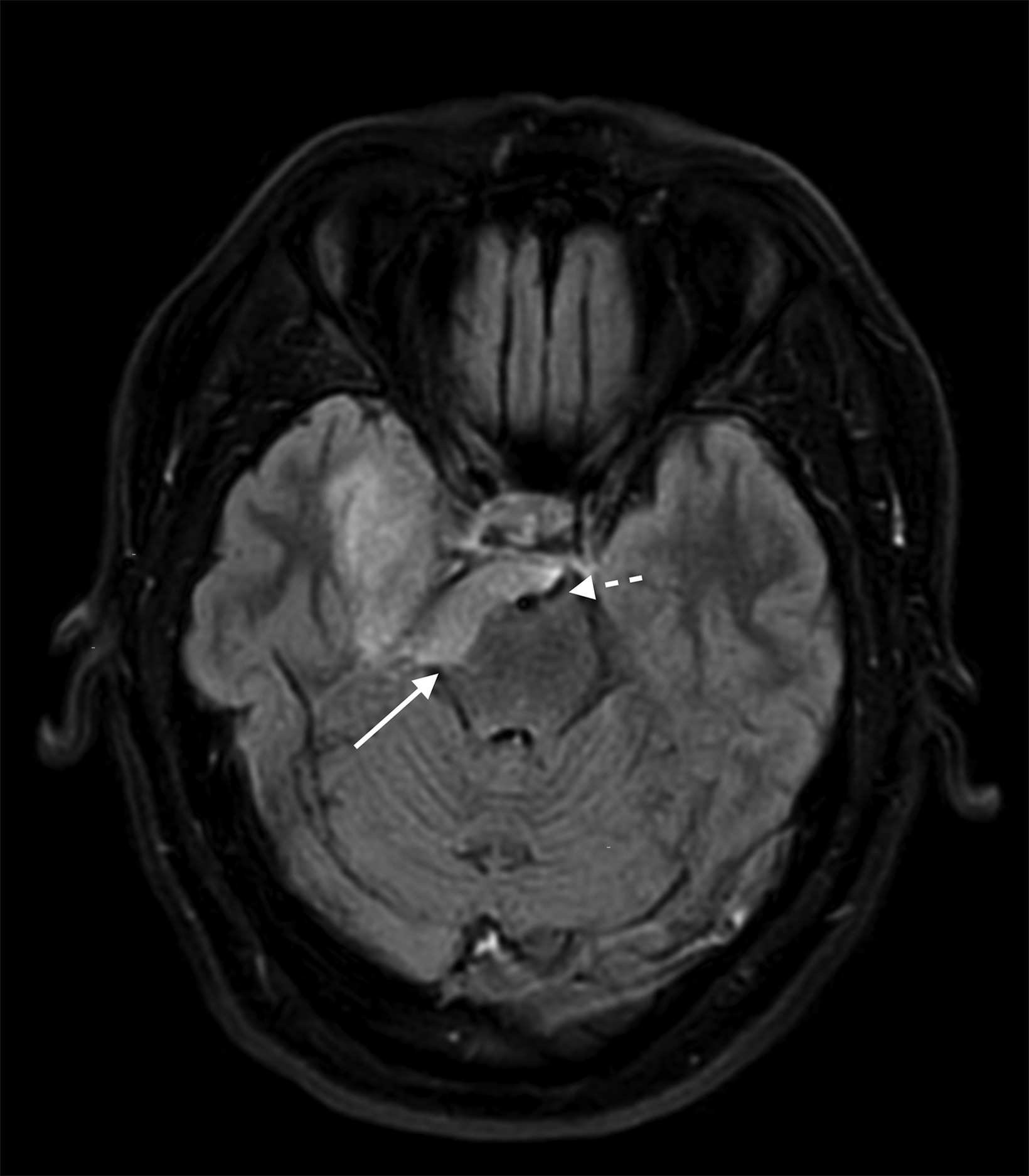
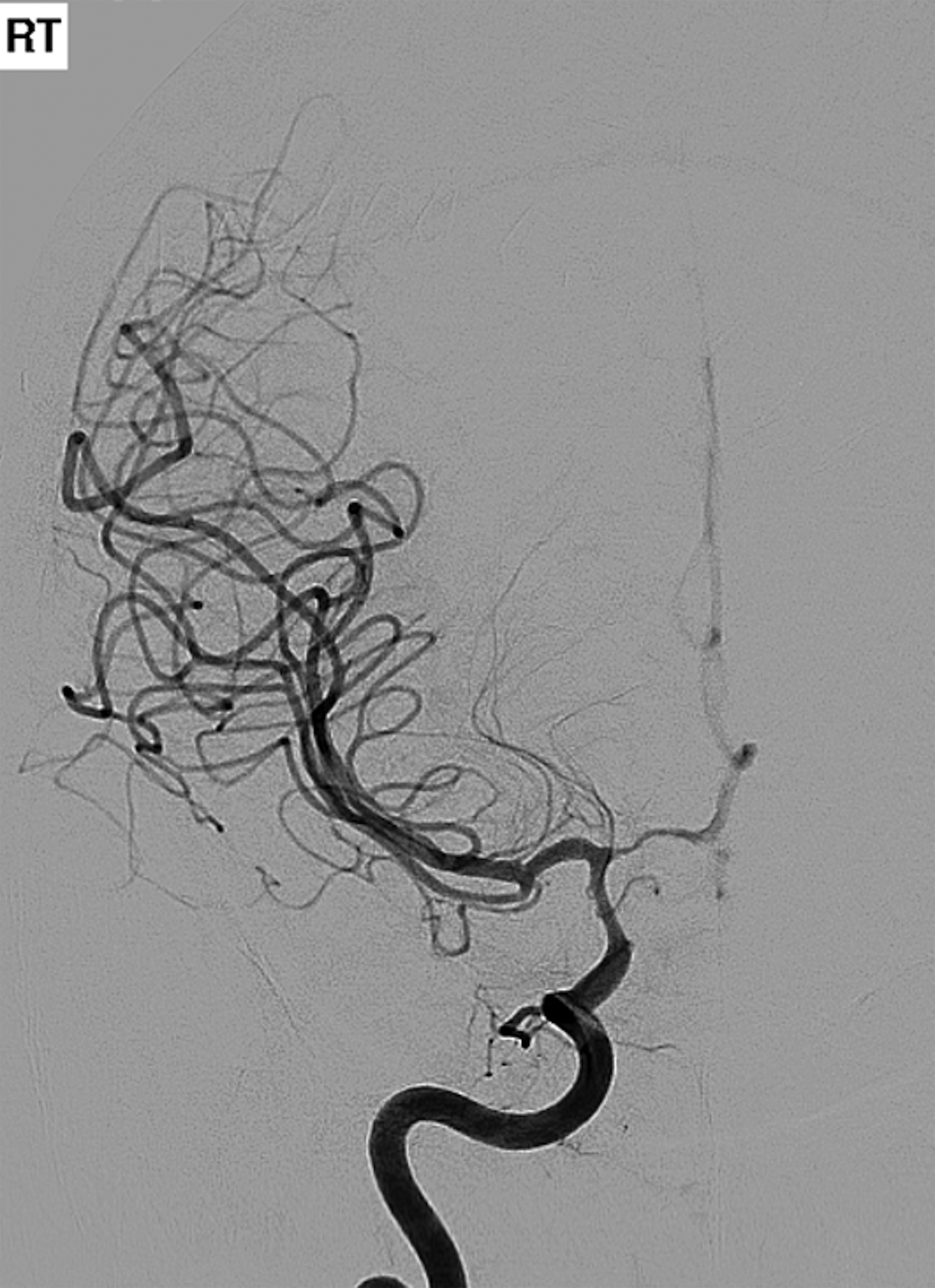
Postoperative digital subtraction angiography (DSA, Figure 2) demonstrated cerebral vasospasm most prominent in the right anterior circulation. Diffusion-weighted imaging (DWI) demonstrated multifocal, right cerebral hemisphere infarcts.
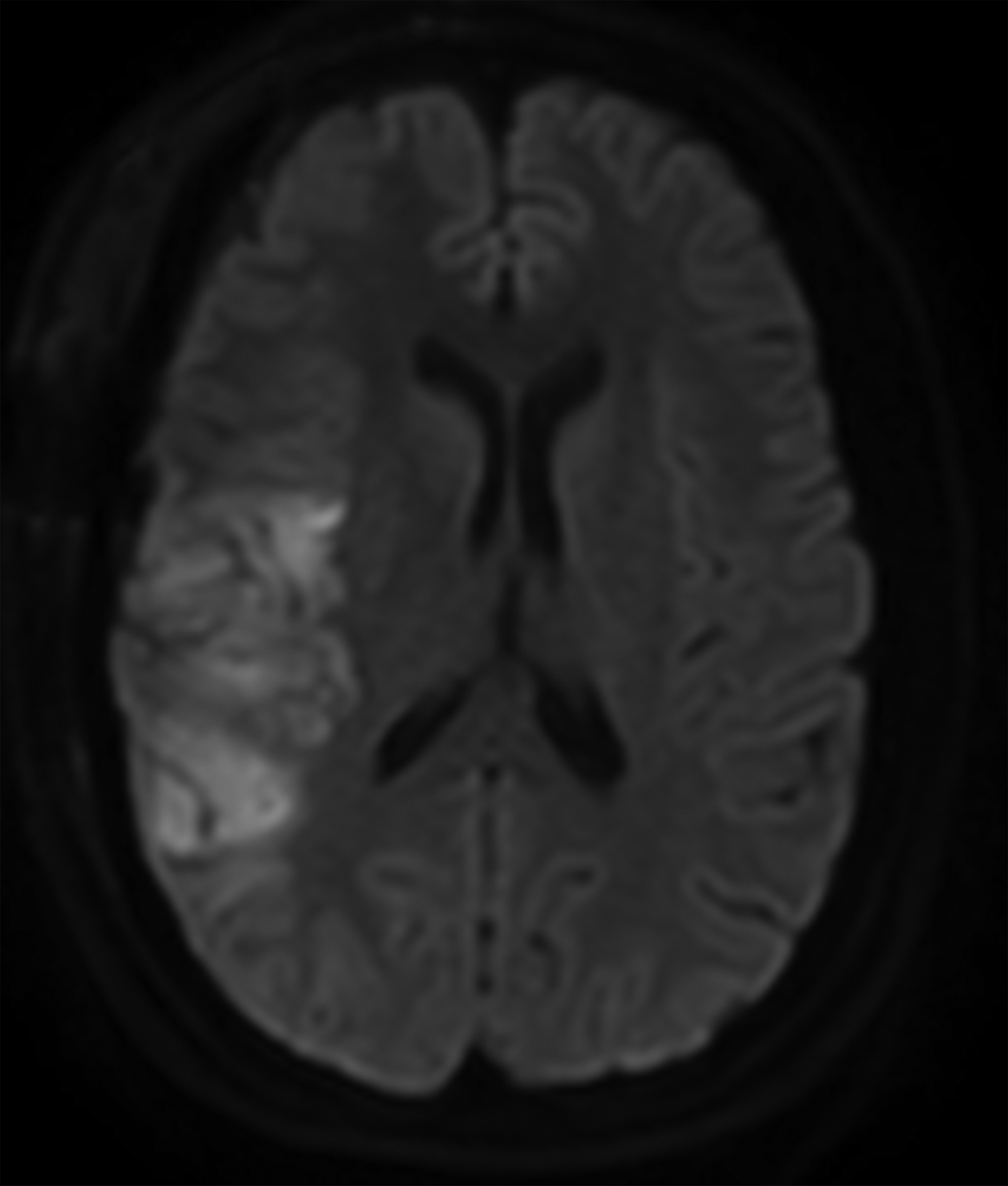
Two months after surgery, axial FLAIR MRI imaging (Figure 3) revealed progression of disease with new tumor extending caudally into the pre-pontine cistern.
Diagnosis
Sellar-subtype atypical teratoid rhabdoid tumor (ATRT), WHO grade IV
Discussion
There is a broad differential diagnosis for sellar/parasellar masses, including benign and malignant neoplasms, as well as numerous nonneoplastic etiologies.1 Primary differential consideration is meningioma. As the most common extra-axial central nervous system (CNS) tumor, meningiomas may occur anywhere dura is present, including in the sellar/parasellar region. A broad, “dural tail” is one characteristic imaging feature of a meningioma. Typical meningiomas are of low grade and grow slowly. However, some high-grade and atypical meningiomas can be locally aggressive and show rapid growth.2
Atypical teratoid rhabdoid tumor (ATRT) is a rare malignant tumor of childhood, constituting approximately 1% of all pediatric brain tumors, but 10-20% of those occurring in children under three years. They are typically seen as intra-axial masses in young children.3,4 These are highly aggressive tumors with substantial potential to metastasize within and beyond the CNS.5 ATRT demonstrates variable imaging features but typically presents as a large, heterogeneously enhancing, intra-axial mass with mixed solid and cystic components related to varying degrees of necrosis, hemorrhage, and/or calcifications.5 The solid components typically show
restricted diffusion on diffusion imaging.
Histologically, ATRTs are composed of rhabdoid cells with vacuolated cytoplasm and mesenchymal spindle-shaped tumor cells.4,5 Varying degrees of primitive neuroectodermal tumor (PNET) cells can create a diagnostic dilemma in differentiating ATRTs from PNET.5 More recently, immunohistochemical analysis has been used to differentiate and characterize ATRT. The new WHO classification defines ATRT as loss of either integrase-interactor 1 (INI-1), tumor suppression gene, or loss of Brahma-related gene 1 (BRG1) protein. Tumors that share histological resemblance but do not harbor genetic alterations are characterized as CNS embryonal tumors with rhabdoid features.6
The sellar subtype of ATRT is an extremely rare variant found almost exclusively in young adult women and, in contrast to the more common pediatric ATRT, is extra-axial and occurs characteristically in a sellar location. Approximately 50 case reports have been published in English literature.3,4,7 On imaging, the tumors have been described as enhancing extra-axial lesions located within the parasellar region. Peripherally located cysts may be present.7 These tumors are aggressive, with mean survival of 23 months. The most common presenting clinical symptoms relate to local mass effect, which includes blurry vision and hypopituitarism features.7 Most tumors recur following resection.
Nakata, et al, described sellar ATRT as a distinct clinicopathological entity occurring exclusively in adult women in the sellar region. They also found different INI-1 alterations compared to classic pediatric ATRT. A characteristic histologic vascular pattern of hemangiopericytoma-like, stag-horn vasculature is thought to distinguish sellar ATRT.4
Pathology in our case showed loss of INI-1, confirming the diagnosis of ATRT. The patient developed vasospasm postoperatively. While vasospasm may be related to recent surgery, vasospasm related to the underlying tumor is also a possibility. Siddiqui, et al, reported a pathological confirmed case of sellar ATRT in a 55-year-old woman presenting with 1 week of headache and blurred vision. The patient was found to have a suprasellar mass with subarachnoid hemorrhage and cerebral vasospasm preoperatively.7
Conclusion
Sellar subtype atypical teratoid rhabdoid tumors (ATRT) are rare, aggressive, WHO grade IV tumors. Sellar ATRT likely represents a distinct clinicopathological entity occurring almost exclusively in young adult women with somewhat differing imaging features and histologic and mutation patterns from typical ATRT.
References
- Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am. 1999;28(1):81-117.
- Kane AJ, Sughrue ME, Rutkowski MJ, et al. Anatomic location is a risk factor for atypical and malignant meningiomas. Cancer. 2011;117(6):1272-1278.
- Shitara S, Akiyama Y. Atypical teratoid/rhabdoid tumor in sellar turcica in an adult: A case report and review of the literature. Surgical Neurology International. 2014;5.
- Nakata S, Nobusawa S, Hirose T, et al. Sellar atypical teratoid/rhabdoid tumor (ATRT). Am J Surg Pathol. 2017;41(7):932-940.
- Meyers SP, Khademian ZP, Biegel JA, Chuang SH, Korones DN, Zimmerman RA. Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. Am J Neuroradiol. 2006;27(5):962-971.
- Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016;131(6):803-820.
- Siddiqui M, Thoms D, Samples D, et al. Atypical teratoid/rhabdoid tumor presenting with subarachnoid and intraventricular hemorrhage. Surgical Neurology International. 2019;10. doi: 10.25259/SNI-59-2019.
References
Citation
R M, J H.Sellar Atypical Teratoid Rhabdoid Tumor. Appl Radiol. 2022; (4):52-54.
July 14, 2022