Radiological Case: Osteoblastic osteosarcoma
Images



CASE SUMMARY
A 43-year-old woman from Puerto Rico presented to a local medical facility with a 2-year history of a left-sided distal forearm mass. The patient denied any history of pain, trauma or constitutional symptoms. Forearm radiographs (Figure 1A) and subsequent contrast-enhanced MRI were performed in Puerto Rico, which raised concern for a bone tumor. An open biopsy was obtained. Histopathology demonstrated irregular lamellar bone with hypocellular fibrous stroma, but no obvious atypia, mitotic activity or necrosis mimicking fibrous dysplasia (Figure 3A). Thus, a benign fibro-osseous lesion was diagnosed and follow-up was recommended. Three months later, follow-up radiographs demonstrated changes consistent with a more aggressive neoplasm. At this point, the patient presented to our institution reporting recent growth and new intermittent dull, burning pain exacerbated by maximal supination. On examination, a firm, nontender mass was appreciated on the dorso-medial aspect of the distal ulna with terminal supination defect compared with the contralateral forearm. Three-view radiographs of the wrist (Figure 1B), high-resolution MRI (Figure 2), and secondary review of outside pathology slides (Figure 3B) were performed.
IMAGING FINDINGS
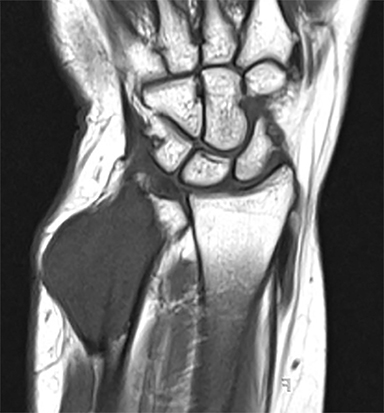
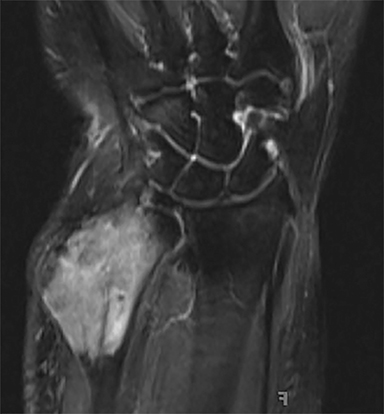
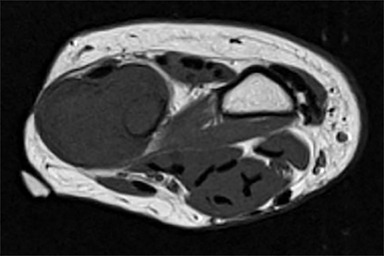
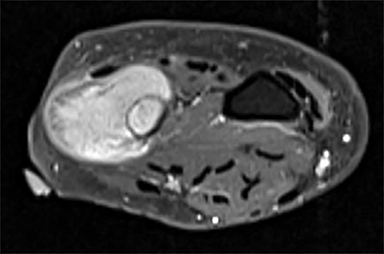
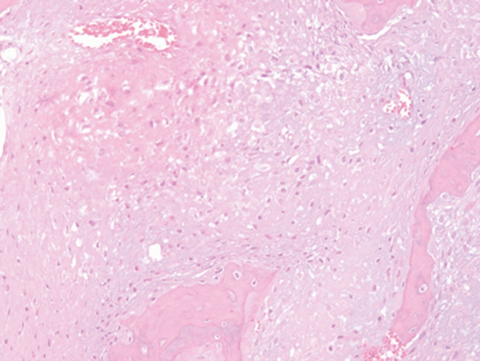
Outside radiographs of the left forearm demonstrated a well-defined, diffusely sclerotic lesion of the distal ulna with a smoothly marginated, radiodense extra-osseous component and notable absence of irregular or interrupted periosteal reaction (Figure 1A). Outside MRI confirmed the presence of a distal ulnar lesion and biopsy was performed, leading to the original diagnosis of a benign fibro-osseous lesion. Follow-up radiographs demonstrated a sclerotic lesion of the distal ulna with an increasing extra-osseous soft tissue mass and new irregular, interrupted, and spiculated periosteal reaction measuring approximately 5 x 2 cm (Figure 1B). High-resolution MRI of the distal ulna was performed to assess internal tumor composition and evaluate local extent of disease (Figure 2). The mass was isointense to skeletal muscle on T1-weighted imaging and moderately hyperintense on T2-weighted imaging with diffuse avid IV contrast enhancement and restricted diffusivity. The mass was centered on the distal ulnar meta-diaphysis with extension to the epiphysis and an extra-osseous soft tissue mass elevating the extensor carpi ulnaris tendon without neurovascular involvement. Review of pathology slides from outside biopsy revealed morphologic features similar to fibrous dysplasia (Figure 3A); however, on closer inspection at high power, malignant cells with round and spindle nuclei were identified infiltrating irregular bone trabeculae with osteoid formation (Figure 3B).
DIAGNOSIS
Osteoblastic osteosarcoma occurring at an atypical age, in an extremely rare location, and with an uncommon radiographic appearance at initial presentation.
DISCUSSION
Beyond childhood/adolescence and in the absence of common imaging findings, failure to consider osteosarcoma can delay diagnosis and negatively impact patient outcomes. Therefore, it is important to recognize that osteosarcoma can occur at any age and at any anatomic site with variable radiographic appearances. Here, we report a case of a 43-year-old female initially presenting with a well-defined, smoothly marginated, purely sclerotic lesion of the distal ulna first diagnosed as a benign fibro-osseous lesion. Uncommon age of presentation, atypical radiographic appearance, and unusual site of disease, as seen in this case, are factors that may contribute to misdiagnosis. Knowledge of common clinical and radiographic findings as well as awareness of atypical presentations should improve reader confidence. We discuss relevant imaging findings, differential diagnostic considerations, and treatment options of osteosarcoma.
Even with a seemingly small incidence of 2 cases per million per year, osteosarcoma is the second-most common primary osseous malignancy behind multiple myeloma. In the 1st and 2nd decades of life, it is the most common primary malignant neoplasm of bone, but osteosarcoma is not uncommon in the third decade of life.1 Osteosarcoma occurring in the 4th or 5th decades as in this case is very rare, but there is a bimodal distribution with increased incidence in elderly patients, typically in the setting of Paget’s disease, osteonecrosis or prior irradiation therapy. Osteosarcoma most often occurs in the metaphysis of long bones, most commonly in the distal femur, followed by the proximal tibia and proximal humerus; less common sites of disease include the skull, pelvis, and jaw.1 A 2007 report of 11,961 cases of osteosarcoma from the National Cancer Database reported by Damron, Ward, and Stewart showed that 57.6% of osteosarcomas occur in the long bones of the lower limb.2 The distal ulna remains an extremely rare site of disease. A large series of 1649 pediatric osteosarcomas published by Arndt and Crist included only two cases of osteosarcomas involving the distal ulna.3
By definition, osteosarcoma is an osteoid-forming spindle cell neoplasm with several histologic subtypes demonstrating variable radiographic appearances, intrinsic cellular heterogeneity, and overall prognosis. Types (histologic subtypes) of osteosarcoma include intramedullary (osteoblastic, chondroblastic, fibroblastic, small cell, telangiectatic, high-grade, low-grade central), surface (parosteal, periosteal, high-grade), and extraskeletal variants.4 The World Health Organizations considers osteoblastic, chondroblastic and fibroblastic subtypes of conventional intramedullary osteosarcoma, the most common type of osteosarcoma. Osteoblastic osteosarcoma, the most common subtype, is diagnosed in the setting of abundant extracellular osteoid production. The matrix comprised of immature bone is deposited in a lace-like pattern with malignant tumor cells incorporated throughout the extracellular matrix.5
Osteoblastic osteosarcoma classically presents as an ill-defined sclerotic, radiodense bony lesion in young patients. However, a mixed sclerotic and osteolytic appearance is more common in practice than a purely sclerotic pattern of disease. Additionally, irregular, interrupted, or complex periosteal reaction including Codman’s triangle, “hair-on-end” appearance, or a spiculated “sunburst” pattern of periosteal reaction is typically present, yet notably absent at the time of initial presentation in this case. Codman’s triangle is defined as elevated and interrupted periosteum forming an acute angle with the underlying cortex due to rapid extension of neoplasm through cortical bone and periosteum into the surrounding soft tissues.5 An accompanying soft tissue mass can often be identified, but usually better demonstrated by cross-sectional imaging such as computed tomography (CT) or magnetic resonance imaging (MRI). In this case, uncommon clinical and radiographic findings at presentation, including a well-defined smoothly marginated purely sclerotic lesion in a middle aged patient without complex periosteal reaction in an extremely rare location, may have contributed to diagnostic uncertainty. Certainly, morphological similarities between the outside biopsy and fibrous dysplasia factored into the original diagnosis of a benign fibro-osseous lesion. In fact, histological differentiation between fibrous dysplasia and low-grade central osteosarcoma can be difficult and may only be possible in the presence of cortical break-through or tumor expression of MDM2 or CDK4.6
Considering patient age, location, and clinical presentation, differential diagnostic considerations for a sclerotic bone lesion may include indolent infection/chronic osteomyelitis, prior trauma or stress reaction, osteoblastic metastatic disease, and osseous lymphoma. When a mixed lytic and sclerotic, moth-eaten, or permeative appearance is identified with cortical disruption and complex periosteal reaction in a young patient, diagnostic considerations may include osteosarcoma, Ewing’s sarcoma, active osteomyelitis, and Langerhans cell histiocytosis.7 Among subtypes of intramedullary osteosarcoma, osteoblastic osteosarcoma generally demonstrates the greatest degree of sclerosis/osteoid formation, while chondroblastic and fibroblastic osteosarcomas may demonstrate areas of lucency juxtaposed with areas of osteoid production. Chondroblastic osteosarcoma may show stippled, punctate, or linear mineralization at radiography or CT, while fibroblastic osteosarcomas often present as a purely lytic, rather than sclerotic, lesion which must be differentiated from other lytic lesions such as malignant fibrous histiocytoma, fibrosarcoma, and giant cell tumor. Telangiectatic osteosarcoma classically presents as a destructive, osteolytic lesion with interrupted periosteum and internal fluid-fluid levels within a soft tissue mass at cross-sectional imaging which may mimic aneurysmal bone cyst. Surface osteosarcomas occur on the cortical surface within or deep to the periosteum and may largely protrude from (parosteal) or extend circumferentially along the surface (periosteal) of the host bone; high-grade surface osteosarcoma may invade the medullary canal.
Treatment and survival rates of osteosarcoma have dramatically improved over the past few decades with the introduction and advancement of more effective chemotherapies and improved surgical techniques. In the early 1900s, the 5-year survival rate of osteosarcoma has been reported to be as low as 5%, rising to 22% by the 1950s.8
However, with the advent of more efficacious chemotherapy regimens and surgical techniques, survival began to significantly improve by the 1970s. Most recently, a 2012 meta-analysis by Allison et al. reported a 5-year overall survival rate of 60% and a 5-year disease-free survival rate of nearly 50%.9 Better outcomes are realized in patients demonstrating tumor response to neoadjuvant therapy seen as greater than 90% tumor necrosis within the surgically resected specimen following preoperative chemotherapy. In fact, the percentage of tumor necrosis following neoadjuvant therapy is the most important prognostic factor for overall and disease free survival in osteosarcomas.10,11 A common chemotherapy regimen may include a combination of neoadjuvant and adjuvant doxorubicin, cisplatin, and high dose methotrexate for a total of 28-30 weeks, though alternative therapies are available. The current treatment algorithm for localized (nonmetastatic) osteosarcoma of the extremity includes neoadjuvant chemotherapy prior to wide local excision most often with limb-salvage and reconstruction and subsequent adjuvant chemotherapy.
CONCLUSION
Osteosarcoma is a malignant, primary, osteoid-forming spindle cell neoplasm of bone with a typical clinical presentation and radiographic appearance. Several histologic subtypes exist that may present with variable imaging appearances. Interpretation of a bone lesion must be performed in light of patient age and location. Absence of classic imaging findings or atypical patient demographics may contribute to diagnostic uncertainty. Here, we present an interesting case of osteoblastic osteosarcoma occurring in a middle age patient outside of the typical age range arising in an extremely rare anatomic location with an atypical radiographic appearance at initial presentation to highlight the importance of maintaining an appropriate index of suspicion despite highly unusual clinical and radiographic findings.
REFERENCES
- Unni KK, Dahlin DC. Dahlin’s bone tumors: General aspects and data on 11,087 cases. 5th ed. Philadelphia: Lippincott-Raven, 1996.
- Damron TA, Ward WG, Stewart A. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National Cancer Data Base Report. Clin Orthop Relat Res. 2007; 459:40-47.
- Arndt CA, Crist WM. Common musculoskeletal tumors of childhood and adolescence. NEJM New Engl J Med.1999; 341(5):342-352.
- Murphey MD, Robbin MR, McRae GA, et al. The many faces of osteosarcoma. Radiographics.1997; 17(5):1205-1231.
- Klein MJ, Siegal GP. Osteosarcoma: Anatomic and histologic variants. Am J Clin Pathol.2006; 125(4):555-581.
- Fletcher CDM, Bridge J, Hogendoorn PCW, Mertens F, eds. WHO Classification of Tumors of Soft Tissue and Bone. 4th ed. International Agency for Research on Cancer; 2013.
- Picci P. Osteosarcoma (osteogenic sarcoma). Orphanet J Rare Diseases. 2007; 2:6.
- Coventry MB, Dahlin DC. Osteogenic sarcoma; A critical analysis of 430 cases. J Bone Joint Surg Am. 1957; 39-A(4):741-757; discussion, 757-748.
- Allison DC, Carney SC, Ahlmann ER, et al. A meta-analysis of osteosarcoma outcomes in the modern medical era. Sarcoma. 2012;2012:704872. doi: 10.1155/2012/704872.
- Rosen G, Nirenberg A. Neoadjuvant chemotherapy for osteogenic sarcoma: A five year follow-up (T-10) and preliminary report of new studies (T-12). Progress in clinical and biological research. 1985; 201:39-51.
- Bacci G, Ferrari S, Bertoni F, et al. Long-term outcome for patients with nonmetastatic osteosarcoma of the extremity treated at the istituto ortopedico rizzoli according to the istituto ortopedico rizzoli/osteosarcoma-2 protocol: an updated report. J Clin Oncol.2000; 18(24):4016-4027.
Citation
Radiological Case: Osteoblastic osteosarcoma. Appl Radiol.
July 1, 2015