Myelin Oligodendrocyte Glycoprotein Antibody-Associated Meningoencephalitis
Case Summary
A child presented to the emergency department with a 6-week history of intermittent fevers, weakness, upper abdominal pain, emesis, and headaches with photophobia. The patient had recently been treated with antibiotics for strep throat and a urinary tract infection.
On physical examination, the patient had a 38.6℃ fever, severe photophobia, hyperreflexia, and mild imbalance when walking in tandem. Otherwise, the patient was awake, alert, and interactive. A lumbar puncture showed an elevated opening pressure (33 cm H 2 O), cerebrospinal fluid (CSF) pleocytosis (97 cells/mm3 ), and low glucose. Laboratory studies showed an elevated erythrocyte sedimentation rate and C-reactive protein, an elevated white blood cell count (13 × 109 /L), and slight anemia with a low mean corpuscular volume. Urinalysis showed hematuria and glucosuria. Of the CSF antigen and antibody tests performed, the antimyelin oligodendrocyte glycoprotein (MOG) antibody titer was positive at 1:10,000.
Imaging Findings
Brain MRI ( Figure 1 ) showed nonenhancing, increased T2 signal abnormality involving the basal ganglia (most prominently the heads of the caudate nuclei), left thalamus, brainstem, and left inferior corona radiata.
Axial FLAIR MRI of the brain shows multiple foci of increased signal within the left middle cerebellar peduncle (A, arrow) and cerebral peduncles (B, arrows); right and left caudate nuclei (C, arrowheads); left posterior thalamus (dashed arrow); right globus pallidus and bilateral putamina.
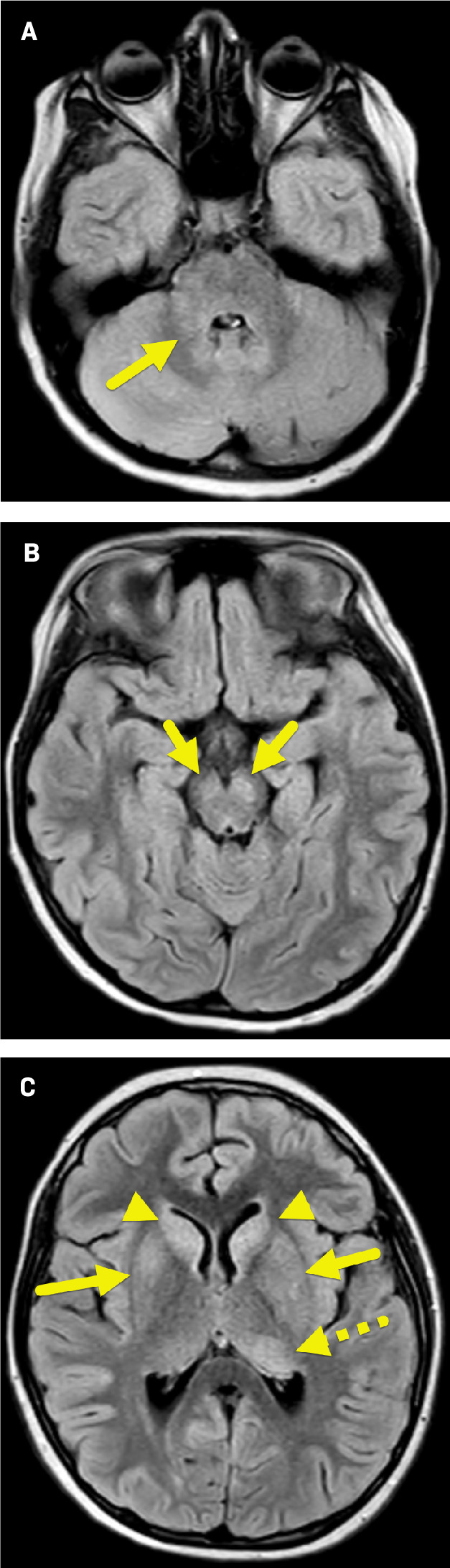
The MRI findings were suggestive of encephalitis. However, because the patient was not encephalopathic and had a wide range of nonspecific subacute symptoms, MOG antibody-associated disease (MOGAD) was suspected.
Diagnosis
Myelin oligodendrocyte glycoprotein antibody-associated meningoencephalitis.
The differential diagnosis for this presentation and MRI findings is broad and includes neuromyelitis optica spectrum disorder (NMOSD), infectious meningitis, acute disseminated encephalomyelitis (ADEM), and multiple sclerosis (MS).
Discussion
Myelin oligodendrocyte glycoprotein antibody-associated disease is an autoimmune disorder that results from the antibodies directed against a myelin glycoprotein found within the central nervous system (CNS).1 Like many autoimmune disorders, the clinical presentation of MOGAD varies depending on the areas of the CNS that are involved. One of the most common presenting symptoms is visual difficulty from optic neuritis.2, 3 The antibodies can attack and cause demyelination in throughout the CNS. When the cerebral hemispheres are affected, an ADEM-like presentation may dominate, while spinal cord involvement may result in signs and symptoms more typical of transverse myelitis.4 Cerebral hemispheric involvement may result in altered mental status and other nonspecific neurological deficits. Alternatively, patients with spinal cord involvement may present with paralysis, sensory disturbances, and bowel and bladder dysfunction.1, 4
The MOG antibody was discovered in 2007 but widespread testing was not available until 2015; therefore, only a relatively small amount of data is available on the incidence and prevalence of MOG encephalitis.1 However, it is thought that children account for approximately 50% of those diagnosed with MOGAD.1, 3 One nationwide Dutch study found the incidence of MOGAD to be approximately 0.16 per 100,000 people.5 In that study, there was a higher seropositivity in children than adults.5 It was also found that up to 56% of children who tested positive for the antibody presented with an ADEM-like illness.5 Other studies have shown that MOGAD occurs more often in White children and does not have a gender predilection.1
In MOGAD, the areas of the brain most often affected are the deep white matter, cortical gray matter with adjacent subcortical white matter, pons, cerebellum, midbrain, medulla, and corpus callosum. The periventricular white matter, area postrema, and deep gray nuclei are affected less frequently ( Figure 1 ).1 It can be challenging to differentiate MOGAD from other inflammatory and demyelinating diseases of the CNS. However, key laboratory, clinical, and imaging features can help narrow the differential diagnosis. For instance, patients with NMOSD often test positive for the astrocytic cell protein APQ4 antibody instead of the MOG antibody.1 Signs, symptoms, and MRI findings for ADEM and MOGAD can appear similar, but ADEM usually follows a monophasic course while MOGAD may relapse.1 While CNS involvement with MS commonly produces relatively more focal brain lesions, the lesions of NMOSD and MOGAD are often larger, even more than 3 cm in diameter.6 Spinal cord lesions have also been reported to span 3 or more continuous vertebral body segments in NMOSD and MOGAD.6 Additionally, serial MRI can be helpful to assess lesion evolution, as lesions are more likely to resolve entirely in patients with MOGAD than in those with MS or NMOSD.6, 7 Multiple sclerosis lesions may decrease in size and/or conspicuity after an attack, but the lesions are often still apparent after signs and symptoms have resolved.6 Follow-up MRI may be particularly useful when the MOG antibody titers are low and there is concern for a false positive diagnosis of MOGAD, allowing one to assess for lesion evolution to support a diagnosis of MOGAD.6
Clinical diagnosis of MOGAD includes the combination of clinical findings suggestive of CNS involvement, testing positive for the MOG antibody by cell-based assay, and exclusion of other conditions.7 Use of CSF sampling and MRI of the orbits, brain, and spinal cord may also aid in diagnosis.1, 3 The CSF of MOG seropositive patients often shows a lymphocytic pleocytosis and a normal to slightly elevated protein level, often without oligoclonal bands.1
Current treatment of MOGAD includes pulsed intravenous methylprednisolone over 3-5 days, followed by a slow oral steroid taper.1 Patients may also be treated with intravenous immunoglobulin (IVIG) or plasma exchange.1 If there are relapsing signs and symptoms, then long-term immunosuppression with rituximab or mycophenolate mofetil may be used.1, 3
The overall prognosis of MOGAD is generally good, and better for children than adults.1 Cobo-Calvo et al2 found that adults were more likely to experience relapse than children (hazard ratio of 1.41). Additionally, using the Expanded Disability Status Scale, adults had more difficulty recovering than children.2 When the degree of MOG antibody titers is elevated and persists, risk for relapse increases.1 A study by de Mol et al5 showed that 89% of patients who became seronegative did not relapse. A different study of 98 children also showed that 64.2% of nonrelapsing children were seronegative for the MOG antibody after 2 years.2 Therefore, a patient’s MOG titer should be rechecked later to determine their risk of repeat CNS attacks and the need for further immunosuppressants.
The patient was treated with pulse dose methylprednisolone for 3 days and a steroid taper. There was no relapse of symptoms.
Conclusion
Myelin oligodendrocyte glycoprotein antibody-associated disease is an autoimmune disorder caused by an antibody directed against MOG in CNS myelin. The clinical presentation varies depending on where inflammation and demyelination occur. Studies have shown that optic neuritis is the most common presenting symptom, but patients may also present with an ADEM-like presentation or transverse myelitis. Myelin oligodendrocyte glycoprotein encephalitis is more common in children than adults, and children are more likely to present similar to ADEM. However, children seem to recover faster and have a lower risk of relapse. Diagnosis is based on clinical presentation of CNS abnormalities, MOG seropositivity, and exclusion of other similar illnesses. Current treatment relies on steroid administration with more aggressive and/or longer--term immunosuppression considered for more severe presentations and relapsed disease.
References
Citation
AA W, Towbin, AJ, MorganD, RB T. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Meningoencephalitis. Appl Radiol. 2024; (4):42 - 44.
doi:10.37549/AR-D-24-0020
August 1, 2024