Imaging pediatric spine tumors
Images



















Spine tumors account for approximately 10 percent of central nervous system (CNS) tumors in the pediatric population. Based on their
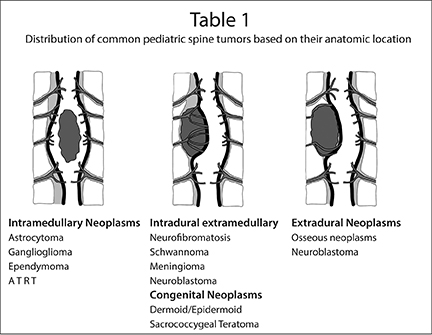
anatomical location, spine tumors of childhood can be classified into intramedullary, intradural/extramedullary and extradural tumors. The vast majority are extradural tumors, accounting for 66% of cases, with intramedullary tumors comprising 25% of cases, and the remainder belonging to the category of intradural/extramedullary tumors (Table 1).1,2 This article discusses the most common pediatric spine tumors, describes their imaging characteristics and reviews the techniques used to image them, primarily contrast-enhanced magnetic reso nance imaging (MRI), but also including computed tomography (CT) in certain cases.
Technical aspects of imaging spine tumors
Magnetic resonance imaging has emerged as the imaging modality of choice for diagnosing and characterizing pediatric spine tumors. With its multi-planar imaging capability, superior contrast resolution, and inherent high signal-to-noise ratio, MRI is best suited to provide information regarding the exact location, extent and morphological features of these tumors. This is crucial in providing a precise diagnosis, or at least narrowing the differential diagnosis. MRI does not involve ionizing radiation, which is particularly important with respect to imaging the pediatric population.
The MRI protocol typically includes unenhanced sagittal T1-, sagittal and axial T2-weighted images, and contrast-enhanced T1-weighted images in the sagittal and axial planes. Coronal images may be particularly useful for intradural/extramedullary tumors and in evaluating tumors with para-spinal extension. Diffusion-weighted imaging may be particularly useful in characterizing high-grade neoplasms and evaluating dermal inclusion cyst/epidermoid tumors. Ongoing work in more advanced MRI techniques, such as diffusion tensor imaging,promises to be a useful tool in characterization of pediatric spinal cord tumors.
Computed tomography (CT) continues to have a role in assessing osseous spinal tumors and detecting calcification/mineralization. Judicious use of CT can be a valuable adjunct to MRI in pediatric spinal tumors.
The clinical presentation of spinal cord tumors may be very nonspecific, with pain or progressive scoliosis. Hence, many children may initially be imaged with conventional radiography.
Clinical presentation of spine tumors
Most spinal cord tumors in children grow slowly. The symptoms may be vague and even misleading; hence, in most cases there is a significant delay in diagnosis. Some common clinical features associated with spinal cord tumors include pain, mild motor weakness, progressive scoliosis and gait disturbances. There may be a history of remission and exacerbation of symptoms thought to be secondary to the varying degrees of peri-tumoral cord edema. Tumors located in the upper cervical spinal cord or at the cranio-cervical junction may present with torticollis and occasionally with lower cranial nerve palsies. Rarely, hydrocephalus and/or raised intracranial pressure can be a manifestation due to altered CSF dynamics produced by the spinal tumor.
Intramedullary tumors
Astrocytoma
Intramedullary tumors comprise approximately 10 percent of all primary CNS tumors, but 25 percent of those in the pediatric population.1,3 Astrocytoma is the most common intramedullary tumor in this group, accounting for up to 60 percent.2,4 Controversy remains regarding the second-most common type of intramedullary tumor—whether it is ganglioglioma or ependymoma. Older literature favors ependymoma, while most of the more recent pediatric literature argues for ganglioglioma, accounting for up to 15%.1,3 Ependymomas of the spinal cord are usually seen in the setting of neurofibromatosis type 2, but are otherwise uncommon in children. The more common myxo-papillary subtype is usually seen in adolescents and is typically intradural/extramedullary in location.5 Other intramedullary cord tumors seen in children include atypical teratoid-rhabdoid tumors, hemangioblastomas and, rarely, intramedullary metastases.1,3,6
The astrocytoma originates from the astrocyte cells of the spinal cord. The most common histological subtype is pilocytic astrocytoma(WHO grade I) followed by fibrillary astrocytoma (WHO grade II).1,5 When increased cellularity, mitosis, and nuclear atypia are present, it is classified as anaplastic astrocytoma (WHO grade III). Rarely, glioblastoma can occur in the spinal cord (WHO grade IV). Multiple low-gradeastrocytomas may be seen in the spinal cord in the setting of neurofibromatosis type 1. Astrocytomas usually affect children between 1 and 5years of age, with the most common astrocytoma being the pilocytic variety.2,5 Fibrillary astrocytoma is more common in older children, usually above 10 years of age.
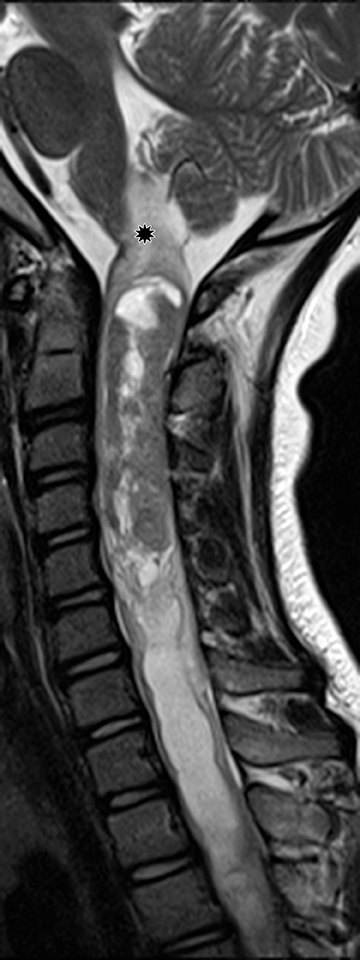
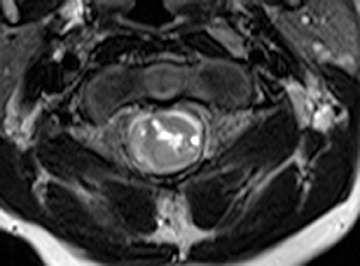
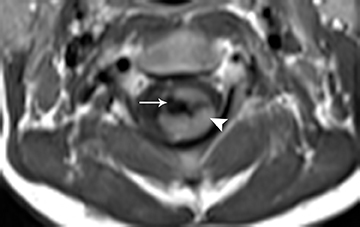
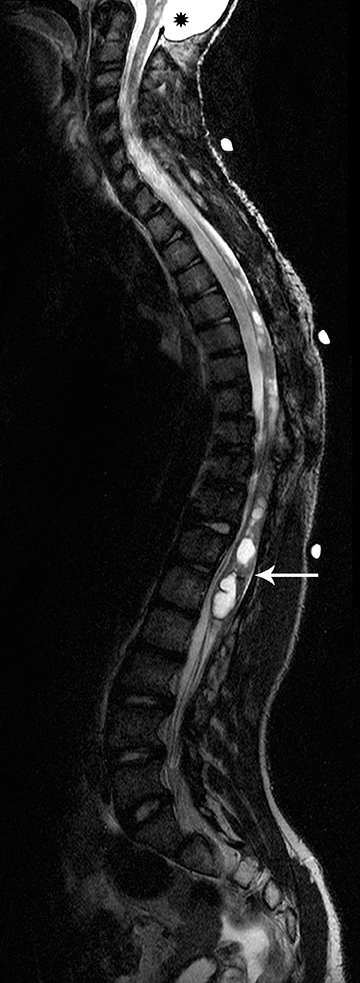
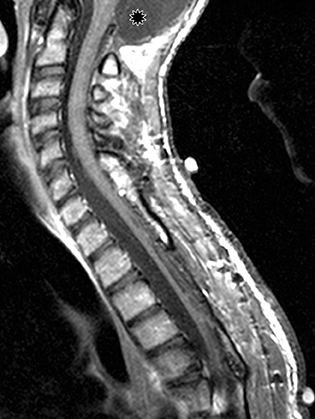
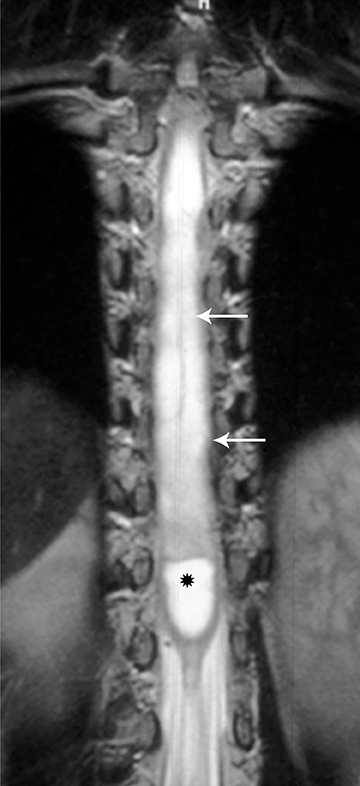
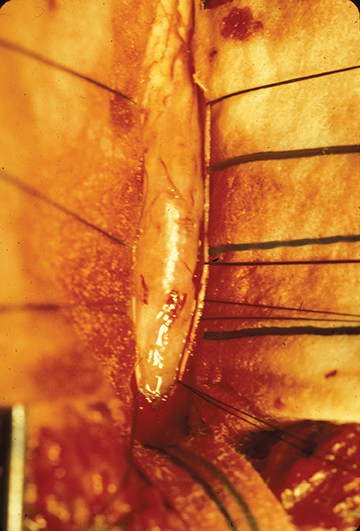
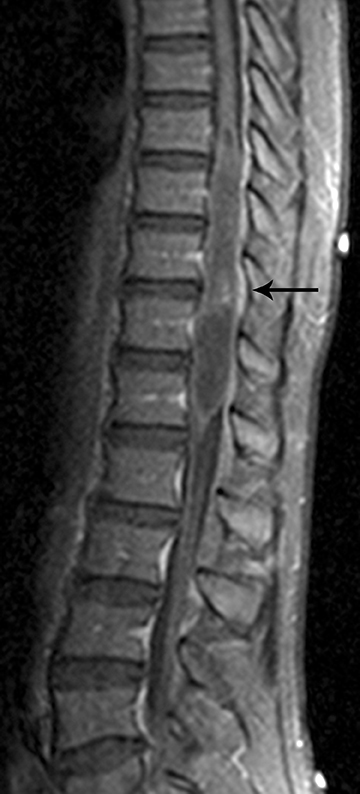
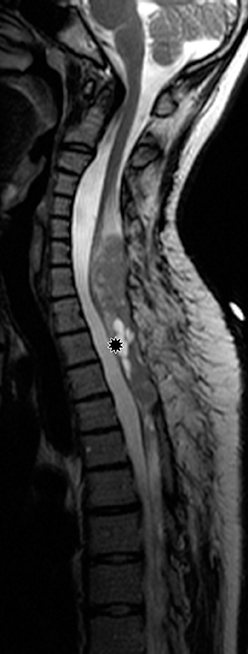
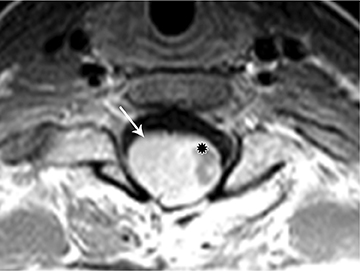
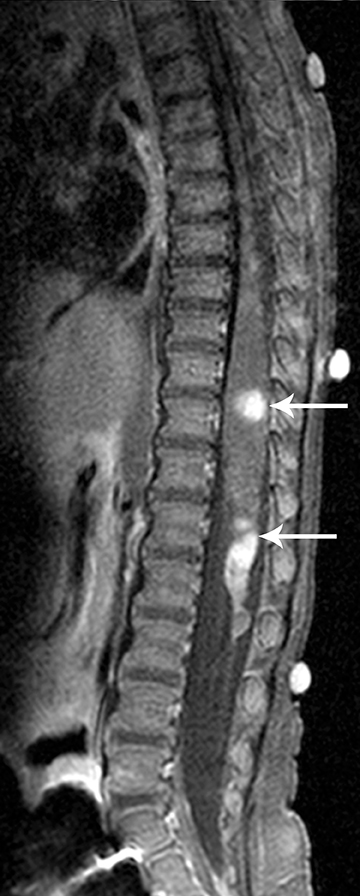
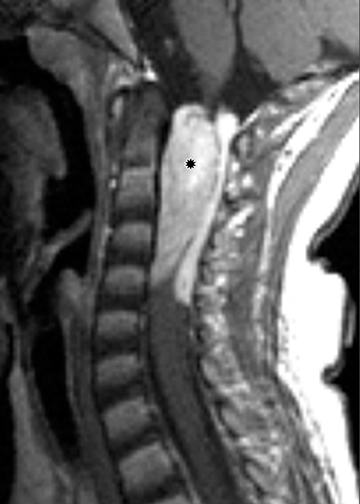
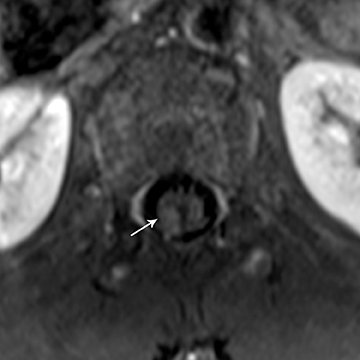
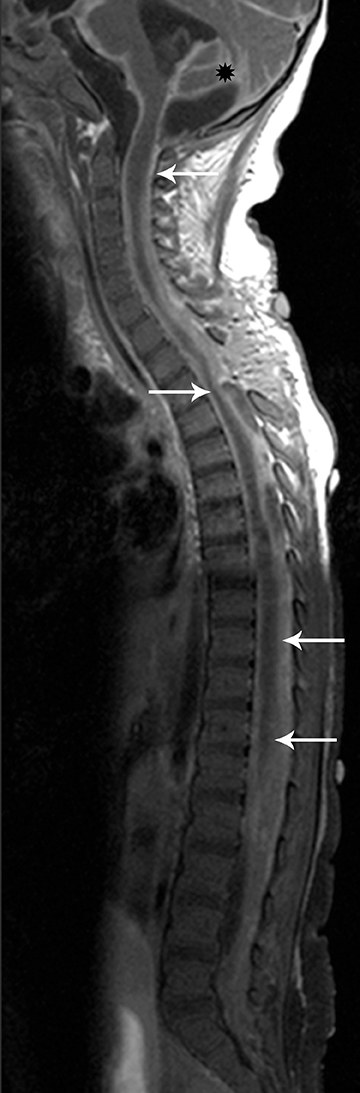
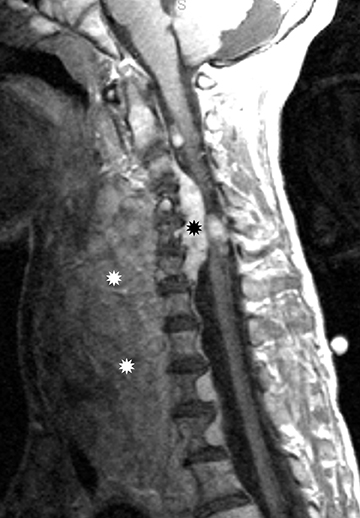
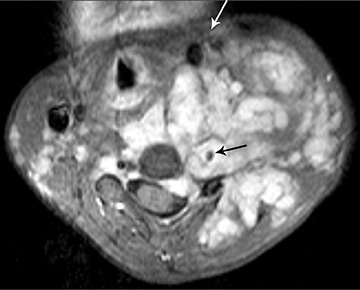
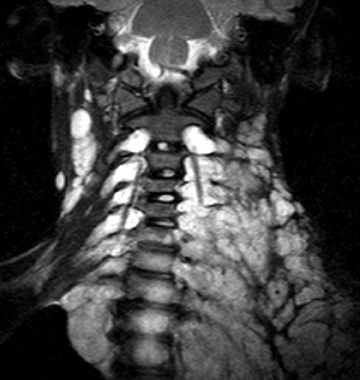
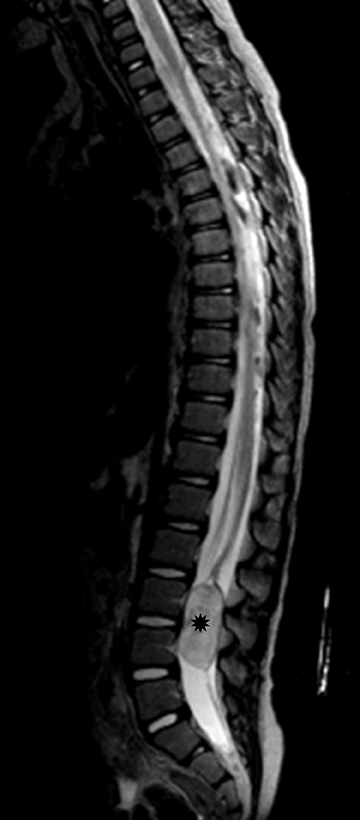
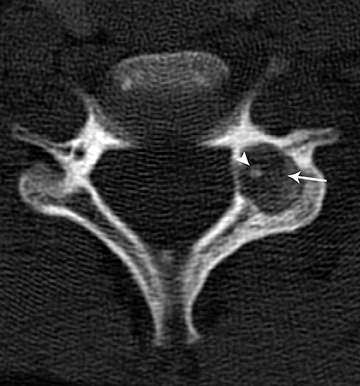
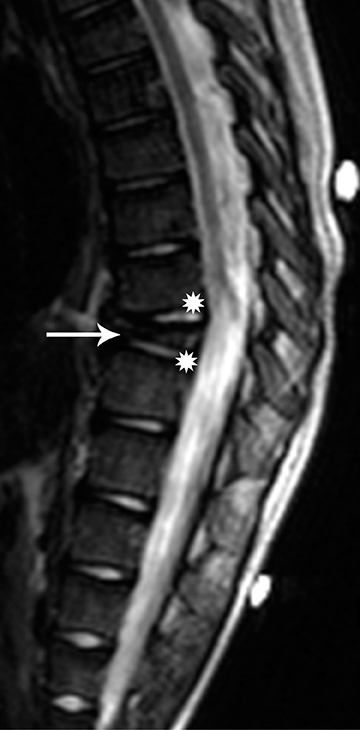
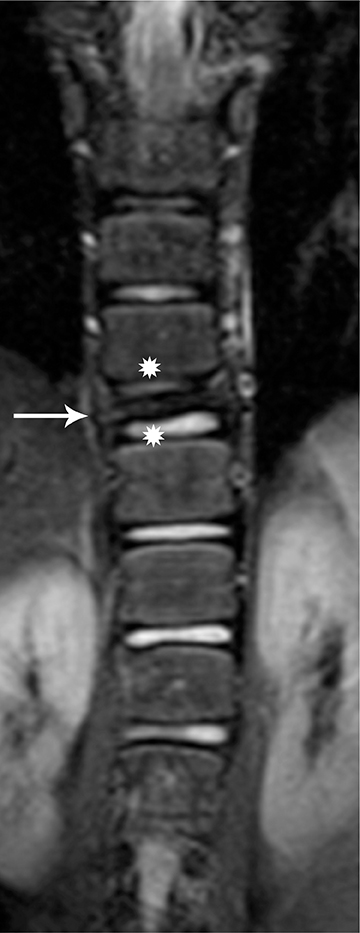
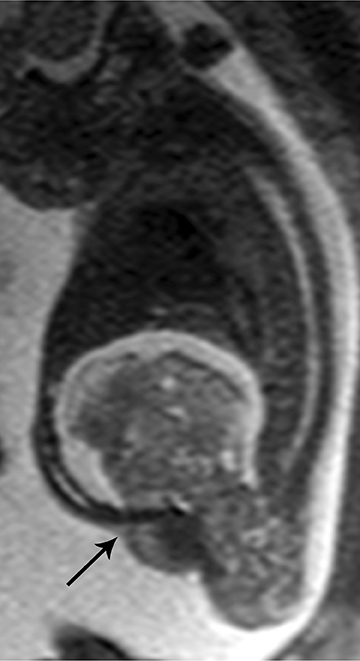
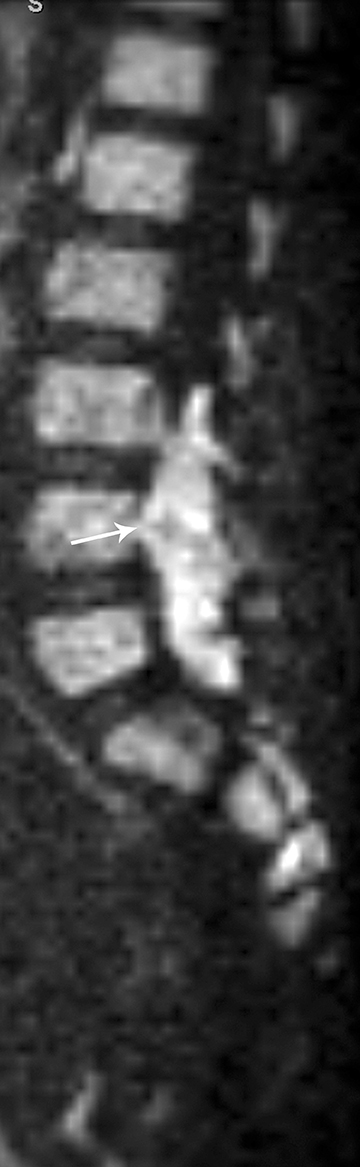
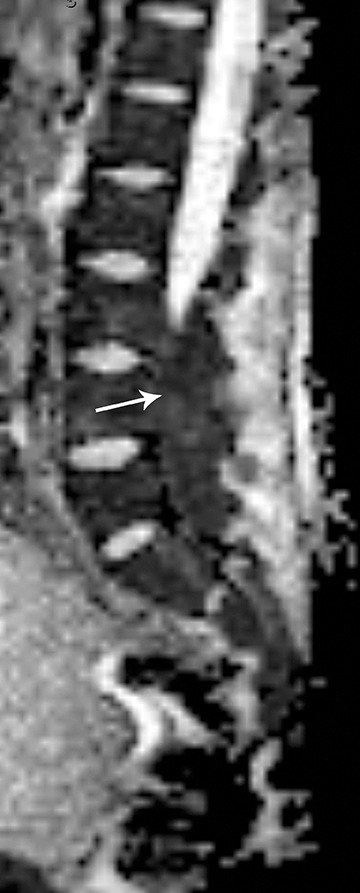
Astrocytomas tend to expand the cord eccentrically (Figure 1). The extent of spinal cord involvement can vary from a few vertebral body segments to almost the entire spinal cord.2 Holocord involvement typically occurs with pilocytic astrocytomas (Figure 2).2 Cysts can occur within the tumor mass itself (Figure 3) or there may be non-neoplastic cranial cysts caudal to the tumor mass and extending for a variable length of the cord.4,6 These non-tumor ‘cysts’ are believed due to dilatation of the central canal (Figure 1).
Eccentric expansion of the spinal cord frequently appears on MRI. Usually astrocytoma is iso-to hypointense on T1-weighted images and shows increased T2 signal. Varying degrees of edema, evident as hyperintense T2 signal, may be present above and below the tumor. Contrast enhancement varies. It has been reported that up to 30 percent of intramedullary astrocytomas do not enhance.2 Those that do enhance most commonly show a heterogeneous pattern. Cysts within the tumor may be seen as rim-enhancing fluid signal areas. The non-tumoral cysts,which are typically seen at the poles of the tumor, do not demonstrate contrast enhancement.
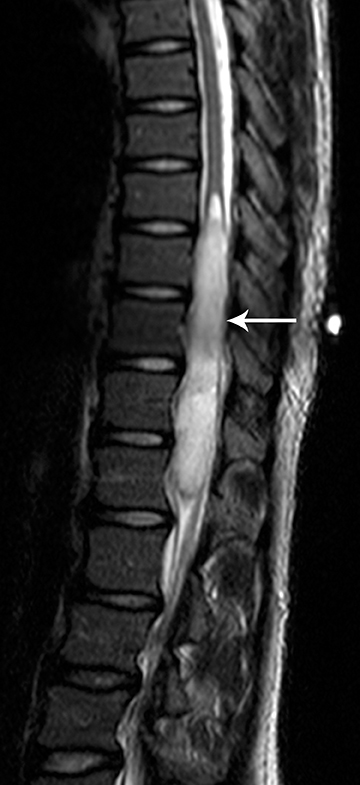
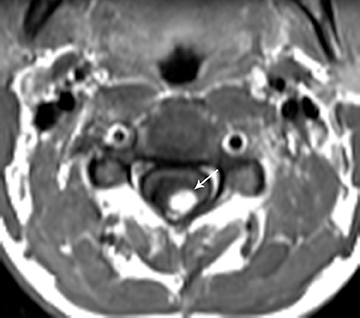
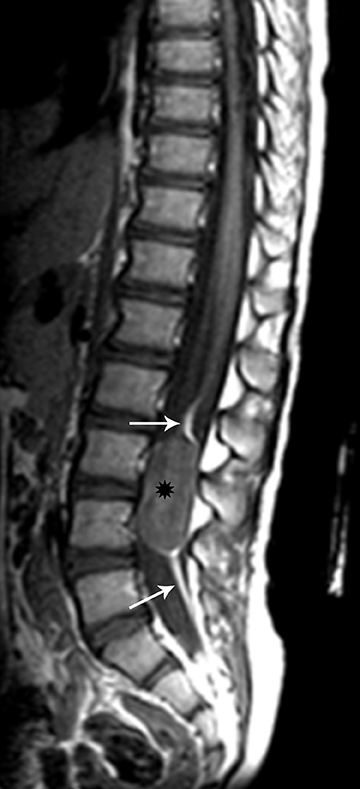
Pilocytic astrocytomas are relatively well circumscribed; however, the higher-grade astrocytomas demonstrate poorly defined margins (Figure 4). Hemorrhage is uncommon, but it may be seen in higher-grade astrocytomas.4 Although rare, CSF dissemination may also be seen with high-grade astrocytomas and has been reported even with pilocytic astrocytomas.4,7 Hence, imaging the entire neural axis with contrast is recommended in all cases of intramedullary astrocytomas.
Often, due to the slow growth and expansion of the spinal cord there may be secondary widening of the spinal canal. Rarely, scalloping of the borders of the adjacent vertebral bodies may be noted.
Surgical excision is the mainstay in treatment of cord astrocytomas. A complete resection is not generally achievable, particularly of those tumors with poorly defined margins. Extensive laminectomies may be necessary, along with postoperative stabilization hardware. The prognosis is most closely related to tumor grade A progression-free, five-year survival rate as high as 95 percent may be attainable for astrocytomas of grade I and II;8 rates drop with higher grades. Patients presenting with neurological dysfunction prior to surgery are at risk for further deterioration.3,9
Ganglioglioma
Histologically, gangliogliomas are composed of neoplastic ganglion cells and glial elements. They occur mostly in children between 1and 5 years of age. Gangliogliomas are usually low——I or II—with low potential for malignant degeneration. However, they are known to have a significant propensity for local recurrence.10 Calcifications may be present, but the incidence of calcification in spinal cord ganglioglioma is less common than in the intracranial ganglioglioma.
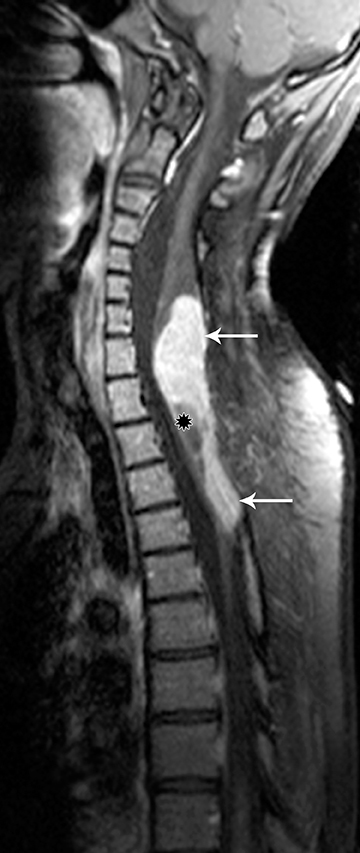
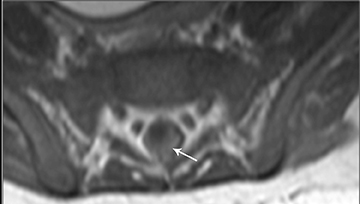
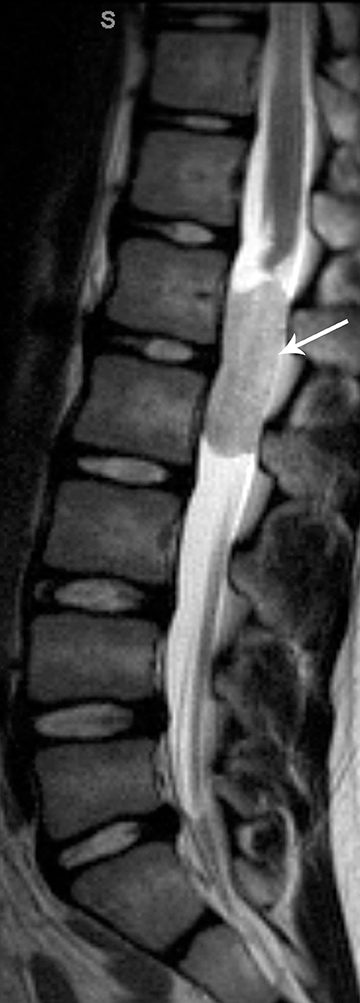
On MRI, the appearance of gangliogliomas is very similar to that of astrocytomas. Imaging findings that favor ganglioglioma include:long segment involvement of the spinal cord, presence of tumoral cysts, and relatively little or no edema proportionate to the size of the lesion.2,11 The signal characteristics also may be more heterogeneous, with mixed signal intensity on T1 and T2 images, thought to be secondary to the dual cellular elements of the tumor and presence of cysts within. Contrast enhancement is irregular and heterogeneous.11 (Figures 5 and 6)
Ependymoma
The intramedullary cord ependymoma in children is usually seen in the setting of neurofibromatosis type 2.2,4,12 Multiple ependymomas may be present. Otherwise intramedullary cord ependymomas are very rare in this age group.
Ependymomas arise from the ependymal cells lining the central canal of the spinal cord; four subtypes have been described: cellular, papillary, clear cell, and tanicytic.4 The cellular form is the most common;4 perivascular pseudo-rosettes are their hallmark histological feature, and most are histologically benign and compress the adjacent cord rather than infiltrate it.2,4On MRI, ependymomas typically appear as well-circumscribed mass lesions causing symmetric central expansion of the cord.2 Due to their compressive rather than infiltrating nature, they usually demonstrate a well-defined margin between the mass and the surrounding cord. Signal within the ependymoma is iso- to- hypo-intense on T1,with variable hyperintensity on T2. Heterogeneity within the ependymoma is common, due to the presence of blood products from intratumoral hemorrhage and/or mineralization. Contrast enhancement is generally present and is usually heterogeneous. A rim of low signal along the border of the neoplasm has been referred to as the “cap sign” and may be seen on T2-weighted images in approximately 20 percent of cases.2,5,12 Intratumoral cysts are usually not a feature of ependymoma, in contrast to astrocytoma and ganglioglioma.2,5 CSF dissemination is rare but maybe seen with high-grade tumors.
Surgical excision is the suggested treatment for spinal cord ependymoma. Prognosis depends on tumor grade, extent of resection, and the presence or absence of CSF dissemination. In many cases, gross total resection is possible due to the presence of a good cleavage margin between the tumor and the cord. With gross total resection the five-year survival rate is approximately 90%.5,13
Atypical teratoid/rhabdoid tumors
Atypical teratoid/rhabdoid tumors (ATRTs) are highly malignant CNS neoplasms that usually occur in children younger than two years.Some of these may be congenital.1 Most ATRTs occur in the brain, but they may also involve the spinal cord. Here they may be either intramedullary or extramedullary in location.1,14 They are aggressive tumors (WHO grade IV) that commonly infiltrate the spinal cord and surrounding structures.
Many of these tumors are very large at presentation, showing aggressive features. On imaging, ATRTs are usually heterogeneous,with solid and cystic/necrotic components, and commonly demonstrate extensive hemorrhage. The solid components demonstrate hypo-intense signal on T1 and hyperintensity on T2.14 Heterogeneous contrast enhancement is commonly seen (Figure 7). CSF dissemination is common; therefore, the entire craniospinal axis should be imaged. The prognosis is uniformly dismal due to the high tumor grade. CSF dissemination and neurological dysfunction are usually noted at presentation.
Hemangioblastoma
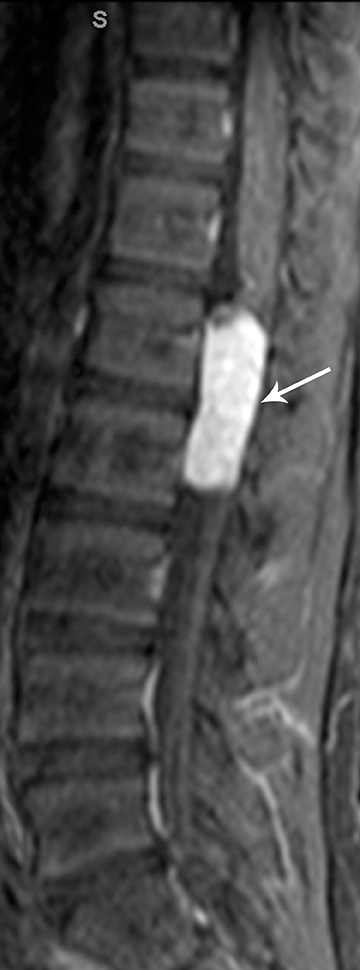
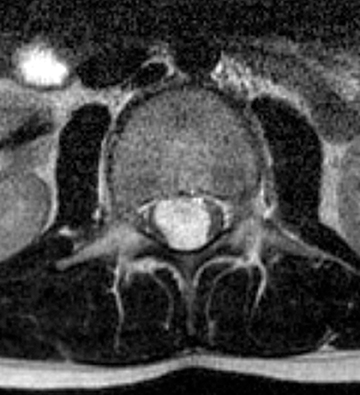
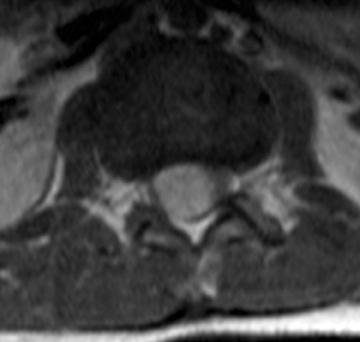
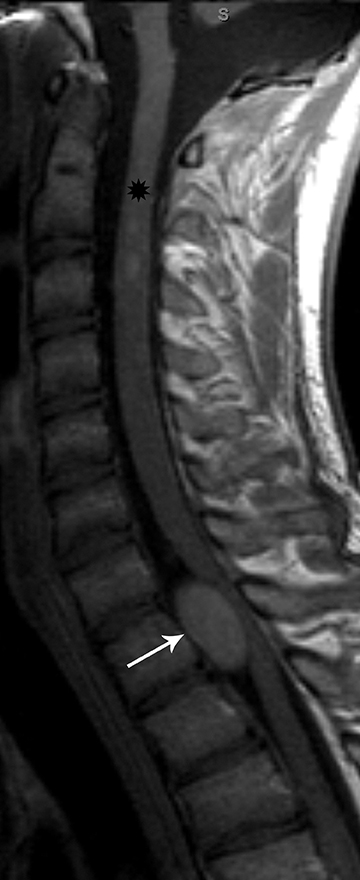
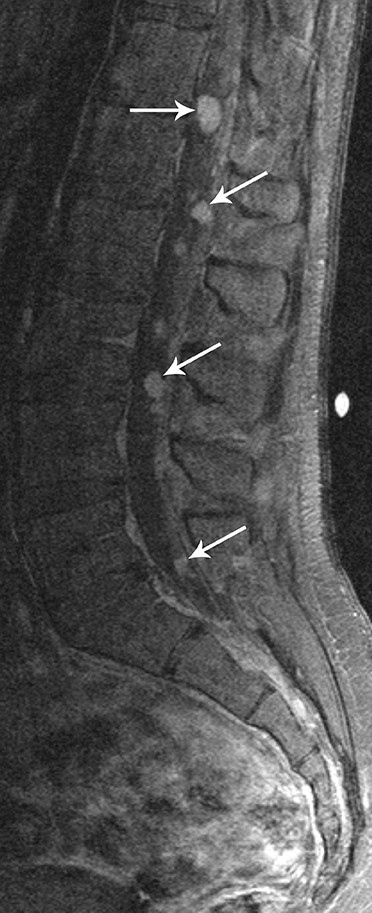
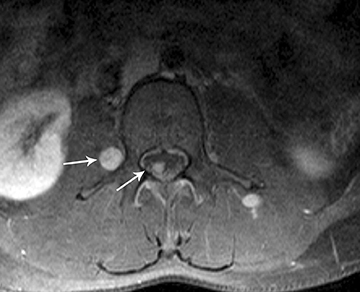
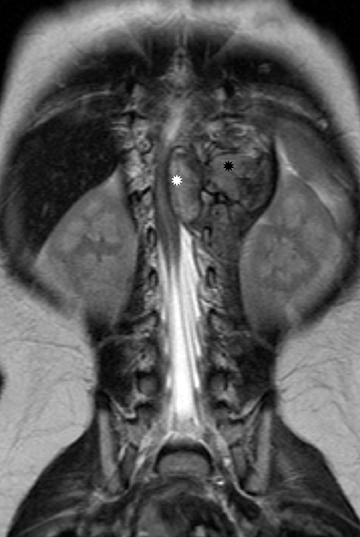
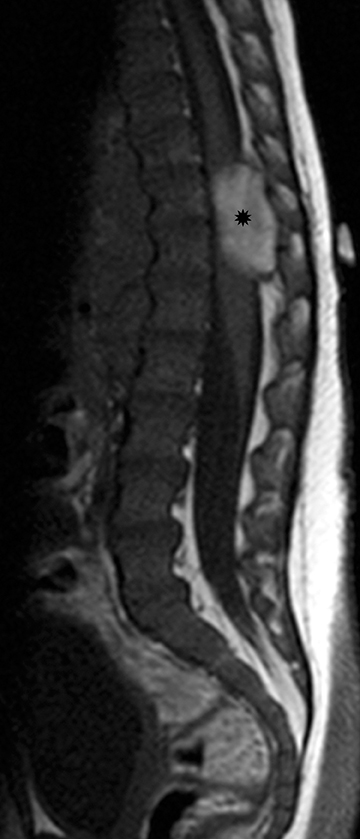
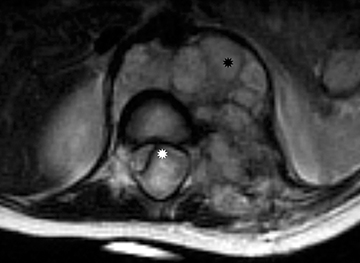
Hemangioblastomas are benign (WHO grade I) but highly vascular, capillary-rich neoplasms. They account for fewer than 10 percent of all spinal tumors and are rare in children.4,5 They can occur sporadically, but are most commonly are seen in children with von Hippel-Lindau(VHL) syndrome, where multiple hemangioblastomas may be present (Figure 8).5 A majority of hemangioblastomas are intramedullary, but they may occur within the intradural space, or even have an extradural localization. Intramedullary hemangioblastomas are peripheral in location, typically arising from the pial surface of the spinal cord.4 On MRI, hemangioblastomas are seen as well-circumscribed nodular masses.The T1 signal may be variable due to the presence of blood products, but they are usually T1 hypo-intense, with hyperintensity on T2 and intense enhancement following contrast administration (Figure 8). There may be varying degrees of surrounding cord edema. In the sporadic cases with single lesions the differential diagnostic considerations may include cavernous malformation, arteriovenous malformation, or other hypervascular spinal cord neoplasms. On angiography, hemangioblastoma is described to have a very prominent vascular blush with a feeding artery and draining vein.1 The treatment of hemangioblastoma is surgical resection. However, due to their extensive vascularity, endovascular embolization often precedes surgery to minimize blood loss.15 Other treatment modalities, such as gamma knife radiosurgery, are also being utilized for the treatment of those hemangioblastomas that are not amenable to surgical resection.16
Clinical presentation
Most spinal cord tumors in children grow slowly. The symptoms may be vague and even misleading; hence, in most cases there is a significant delay in diagnosis. Some common clinical features associated with spinal cord tumors include pain, mild motor weakness, progressive scoliosis and gait disturbances. There may be a history of remission and exacerbation of symptoms thought to be secondary to the varying degrees of peri-tumoral cord edema. Tumors located in the upper cervical spinal cord or at the cranio-cervical junction may present with torticollis and occasionally with lower cranial nerve palsies. Rarely, hydrocephalus and/or raised intracranial pressure can be a manifestation due to altered CSF dynamics produced by the spinal tumor.
Intradural/extramedullary tumors
Tumors in this location characteristically displace and compress the spinal cord and expand the ipsilateral thecal sac. The interface formed between the lesion and the adjacent spinal cord results in the so-called “meniscus sign.”17 These lesions can be located in the neural foramina,the filum terminale or the cauda equina nerve roots. These spinal tumors most commonly present with pain, and, based on the location of the mass, feature spinal-cord or nerve-root compression. The most frequently encountered intradural/extramedullary spinal tumors in the pediatric age group are metastases from primary intracranial tumors.
Other intradural/extramedullary spine tumors in children include the myxo-papillary subgroup of ependymomas, nerve-sheath tumors, such as schwannoma and neurofibroma; congenital or dysontogenetic masses such as the dermal inclusion cyst (dermoid andepidermoid cyst); neurenteric cysts, and arachnoid cysts. Other tumors that may also be found in the intradural/extramedullary location include ATRTs, meningiomas and paragangliomas.12
Metastases
The most common spinal metastases are from posterior fossa tumors, particularly medulloblastoma, followed by ependymoma, among other tumors. Metastases are usually lepto-meningeal in location. On contrast-enhanced MRI the metastases are most commonly seen as diffuse-enhancing over the surface of the cord and nerve roots. The MRI appearance of widespread lepto-meningeal metastases has been described as “sugar-coating” the spinal cord and nerve roots (Figure 9).1 However, many metastases do not enhance with contrast, particularly early in their development, and may be seen only as multiple focal nodular deposits. The absence of contrast enhancement should no tpreclude the diagnosis of lepto-meningeal spread of disease.2
Myxopapillary ependymoma
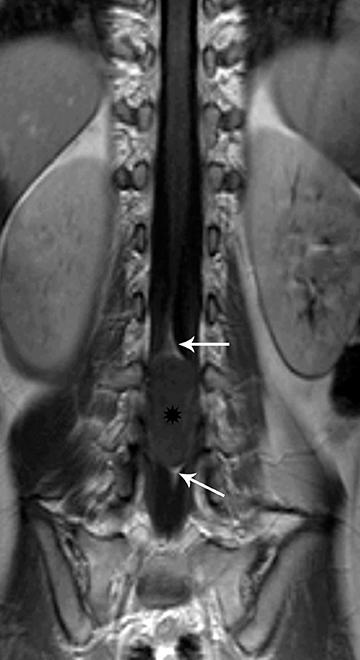
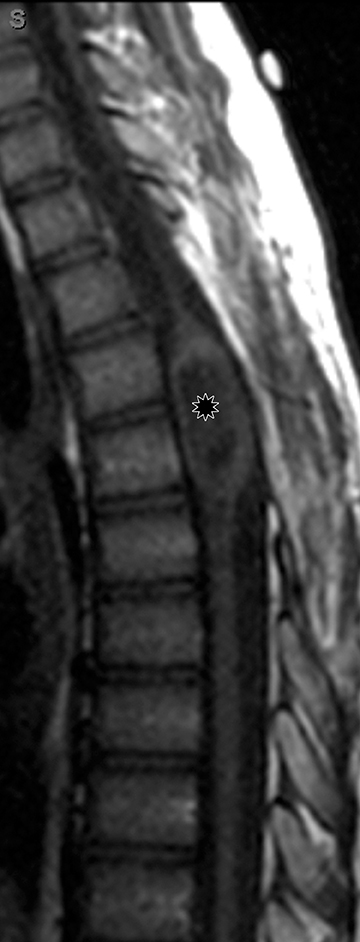
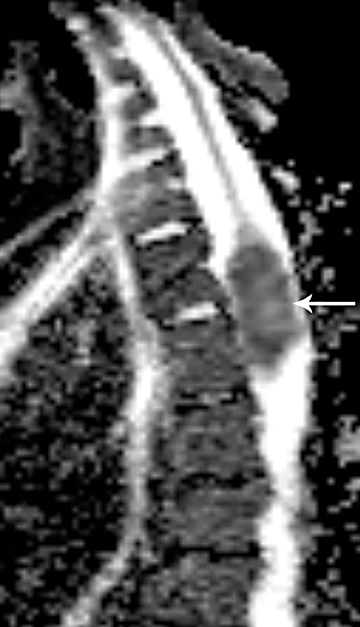
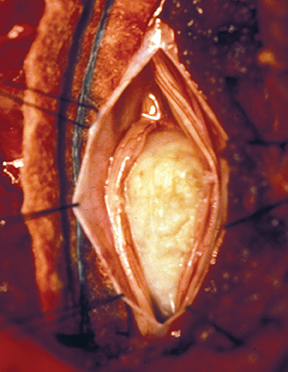
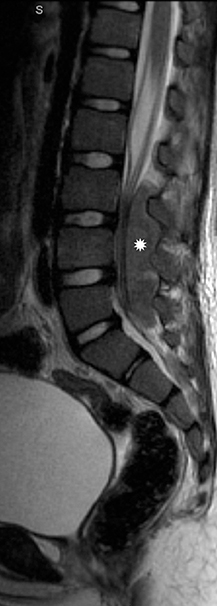
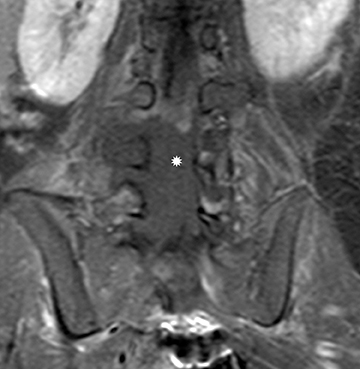
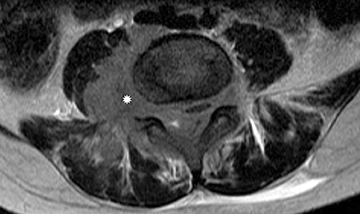
Myxopapillary ependymomas (MPE) are a subtype of ependymoma that most commonly occurs near the conus and filum terminale.18,19 Overall, MPEs account for 13 percent of all ependymomas.5 They arise from the ependymal glial cells and typically present as intradural /extra medullary lesions involving the lumbar and sacral spinal canal.18 The sacro-coccygeal region is another potential site of MPEs, arising from the vestigial cells at the distal portion of the neural tube.5 MPEs are another type of benign tumor (WHO grades I) that can demonstrate CSF dissemination.
Clinical presentation usually includes back or leg pain, lower extremity weakness, and/or sphincter dysfunction. Superficial siderosis may develop associated with repeated hemorrhage from an MPE. Patients may present with sensorineural hearing loss,although this is rare in children.
On MRI, MPEs are iso- to hyperintense on T1-weighted images and heterogeneously hyperintense on T2-weighted images. MPEs are highly vascular and can have intratumoral hemorrhage, accounting for T1 hyper-intensity. T2 heterogeneity is found due to presence of intra-tumoral hemorrhage, mineralization and cysts. Following contrast, MPEs demonstrate considerable patchy enhancement (Figure10).
Nerve sheath tumors
Schwannomas and neurofibromas are common types of nerve sheath tumors. They may be benign, potentially isolated, sporadic lesions or they may be associated with phakomatoses. When isolated, the clinical presentation of schwannomas and neurofibromas is similar to symptoms of radicular pain and short-segment scoliosis.
Schwannomas arise from the Schwann cells in the nerve sheaths of spinal nerves and grow extrinsically with respect to the axon.They may be intradural, extradural or both, depending upon where they originate along the spinal nerve root; however, the intradural location is more common. A thin capsule typically separates the Schwannoma from surrounding tissues.18 Hence, on MRI it appears as a well-defined mass. Schwannomas are iso- to hypointense on T1-weighted images and hyper-intense on T2-weighted images.Avid, homogeneous enhancement is noted following contrast administration (Figure 11). Some large schwannomas may develop areas of cystic degeneration.17 Schwannomas may be solitary and sporadic in the pediatric population. However, they are more commonly associated with neurofibromatosis type 2, and the more recently established entity of schwannomatosis.20
Neurofibromas consist of mixed or Schwann cells and fibroblasts and tend to infiltrate the nerve root from which they originate.18 A clear wall of separation from surrounding tissues is usually lacking, making radical resection difficult. Neurofibromas usually occur in patients with neurofibromatosis type 1. They may be solitary, multiple or form a conglomerate mass termed “plexiform neurofibroma” (Figure 12)..21 Neurofibromas affecting several nerve roots produce an appearance like a “string of beads.”2,21On MRI, the signal characteristics of neurofibromas are similar to that of schwannomas, with iso- to hypointensity on T1-weighted images and hyperintensity on T2-weighted images,with avid contrast enhancement. When the imaging plane is perpendicular to the long axis of the neurofibroma, on T2-weighted or short tau inversion recovery (STIR) images, the typical “target” appearance may be noted.21Solitary schwannomas as well as neurofibromas can extend via the neural foramen into the para-spinal region, producing the characteristic “dumb-bell” configuration.6, 17
Dysontogenetic masses
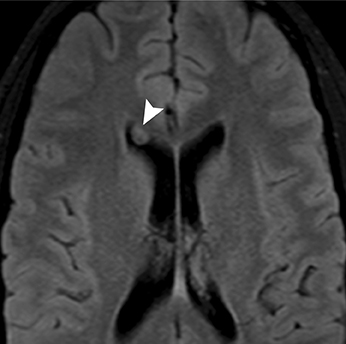
This group includes non-neoplastic masses of congenital origin, such as dermal inclusion cysts (dermoids and epidermoids), neurenteric cysts, and arachnoid cysts. Dermoid and epidermoid tumors account for 10 percent of all spinal tumors in children. Structurally the dermoid has all the three dermal layers and contains dermal appendages; eg, hair, and sebaceous glands. Unlike the dermoid, epidermoid has only two dermal layers and does not contain skin appendages. Both dermoids and epidermoids can have cystic structures and fluid debris with varying concentrations of keratin.18 Dermoids are commonly associated with dermal sinus tracts. Complications such as infection from the dermal sinus tract, meningitis, and abscess formation may occur. The MR signal characteristics of the dermoid can vary depending on the composition of the cystic content and amount of solid tissue. The cystic content on MR may demonstrate intrinsic high T1 signal and intermediate-to-low T2 signal. Solid components of the dermoids are iso-to hypo-intense relative to the spinal cord on T1-weighted images and hyper-intense on T2-weighted images. Typically, dermoids will not enhance with contrast unless secondarily infected (Figure 13).18Epidermoids may be primary or, as is more common in children, iatrogenic secondary to inadvertent inclusion of skin debris during surgical intervention, such as repair of spinal dysraphism. Epidermoids display signal intensity similar to that of CSF on T1- and T2-weighted images.
Diffusion-weighted imaging is extremely helpful in distinguishing an epidermoid from arachnoid cysts due to the characteristic restricted diffusion within an epidermoid.18 Similar to dermoid, epidermoid also demonstrates lack of contrast enhancement, unless secondarily infected (Figure 14).
Extradural tumors
Extradural tumors account for two-thirds of all spinal tumors in children, and may be divided into primary bone tumors, diffuse tumors of the epidural space (such as hematological malignancies) and extra-spinal tumors invading the spine.2 The common benign primary bone tumorsof the pediatric age group include hemangiomas, osteoid osteomas, osteoblastomas, Langerhan cell histiocytosis, aneurysmal bone cysts and sacrococcygeal teratomas. Common malignant bone tumors include Ewing’s sarcoma and osteosarcoma. Lymphomas and leukemias are the most common hematological malignancies of childhood, and they may manifest as diffuse tumors of the epidural space. Primary extra-spinal tumors of the pediatric age group with a propensity for spinal invasion are neuroblastomas, germ cell tumors, and rhabdomyosarcomas. Extramedullary hematopoiesis, such as may be seen in patients with thalasemia may result in multiple large paravertebral or epidural masses that can encroach upon the spinal canal.
Osteoid osteoma and osteoblastoma
Osteoid osteomas and osteoblastomas are benign bone tumors and are considered variants of the same histopathology, differing only in size.22 They may be found in the appendicular skeleton, but the spine is a favored location. Both lesions present with characteristic night pain, relieved with non-steroidal anti-inflammatory drugs. Osteoid osteoma is more common in the posterior elements of the spine. CT is usually the modality of choice for diagnosing osteoid osteomas. On CT, osteoid osteomas can be identified by their radiolucent nidus and surrounding sclerosis. The nidus diameter is usually less than 2 cm and may contain a central calcification (Figure 15). On MRI, osteoidosteomas are hypo-intense on T1-weighted images and heterogeneously hyper-intense on T2-weighted images, with a variable degree of contrast enhancement. Edema surrounding the lesion will be more readily seen with MRI.
Most osteoblastomas arise from the spine. They are usually large at presentation and, due to their heterogeneous appearance, they may mimic malignancy or vascular malformation. Osteoblastomas demonstrate a more aggressive appearance with frequent hemorrhage, cortical disruption, and soft-tissue extension. On CT the nidus of osteoblastoma is usually larger than 2 cm. Osteoblastoma on CT is seen as an expansive lytic lesion, displaying a lower degree of surrounding bony sclerosis. On MRI, the signal is heterogeneous on both T1- and T2-weighted images. Marked contrast enhancement and increased vascularity may also be evident.22
Langerhan cell histiocytosis
Langerhan cell histiocytosis (LCH) is a disorder of the reticular-endothelial system encountered in children and young adults. The disease spectrum includes many pathological conditions, ranging from multisystem disease (Letterer-Siwe and Hand-Schuller-Christian disease) to a single-system disease confined to the skeleton (eosinophilic granuloma-EG).23 EG of the spine has a predilection for the vertebral bodies, particularly in the cervical spine, which is involved in almost 75 percent of cases.1 EG classically causes complete collapse of a vertebral body (“vertebra plana”). The endplates are spared and, hence, remain intact. Radiographs and CT will demonstrate a well-defined osteolytic lesion with varying degrees of vertebral collapse. MRI signal of the collapsed vertebra can be variable but is usually hypo-intense on T1 and hyperintenseon T2, with marked contrast enhancement of the collapsed vertebra and associated soft tissue components (Figure 16). Compensatory swelling of the adjacent intact discs can be seen on MRI. Extension of soft tissue components into the epidural space and spinal cord compression can be better assessed with MRI.23
Sacrococcygeal teratoma
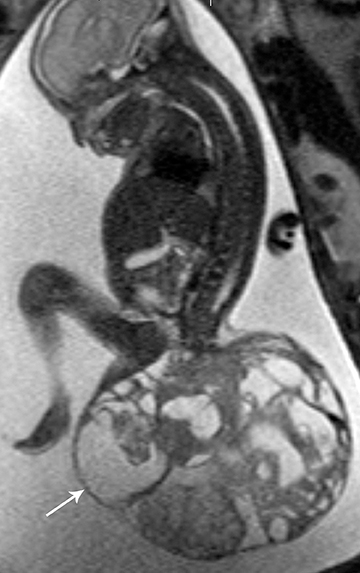
Sacrococcygeal teratoma (SCT) is one of the most common malignancies found in the neonatal age group. Sacrococcygeal teratoma arises from the totipotent cells of the caudal cell mass. Most (about two-thirds) are benign and the remainder are immature or malignant teratomas.24
SCT is characterized by heterogeneous composition including fat, soft tissue, cysts, calcium and hemorrhage. SCT is often large and lobulated at presentation with well-defined margins. Four types have been determined, based on their location: Type I is a predominantly external mass without a significant pre-sacral component; Type II is a large external lesion with a significant intrapelvic component; Type III is a predominantly intrapelvic mass; and Type IV is an intrapelvic mass without an external component.
On MRI, SCTs can have variable signal on T1, depending on their contents, and are usually heterogeneously hyperintense on T2weighted images, with patchy heterogeneous enhancement following contrast administration. Intra-lesional hemorrhage and calcification are common (Figure 17). Antenatal diagnosis of SCT can be established by ultrasound imaging and well characterized with fetal MRI.25 SCT may be present in isolation or may be associated with ano-rectal malformations and caudal regression syndrome in the setting of the Currarino triad.26
Lymphoma and leukemia
Lymphoma and leukemia account for more than 40% of all malignancies affecting the pediatric population. Among these, acute lymphoblastic leukemia (ALL) is the most common variety, and Hodgkin and non-Hodgkin’s lymphoma account equally for the major groups of lymphomas.Spinal involvement is more frequent with non-Hodgkin’s lymphoma. Spinal involvement in both lymphoma and leukemia is frequently manifested as bone marrow infiltration. This may be the only finding or it may be associated with epidural and/or para-spinal soft tissue masses. OnMRI, diffuse marrow infiltration can be seen as abnormally low T1 signal throughout the vertebral bodies and hyperintensity on T2-weighted images, with enhancement on postcontrast imaging.27 Focal mass formation in lymphoma may preferentially involve the epidural space, with avidly contrast-enhancing masses that displace the epidural fat and may cause thecal sac and/or spinal cord compression.
Granulocytic sarcoma (GS) is a localized tumor formed by primitive myeloid cells at an extra medullary site. It is usually associated with myeloid leukemia and is known by other names, such as chloroma, myeloid sarcoma, and extra medullary myeloid tumor. GS is the most common extra-osseous spinal mass in patients with acute myeloid leukemia (AML).28 These are highly vascular solid masses which, onMRI, appear iso- to hyper-intense on T1-weighted images, and intermediate- to hypo-intense on T2-weighted images, and show marked contrast enhancement, as well as diffusion restriction.29 Similar to focal masses of lymphoma, GS can cause effacement of the thecal sac and cord compression (Figure 18).
Extra-spinal tumors with spinal invasion
Neuroblastoma is the third-most common tumor in children after lympho-reticular malignancies, and CNS tumors, and it generally affects children younger than 5 years.2, 30 Neuroblastoma arises from neural crest elements in the adrenal glands, the paravertebral sympathetic chains, the organ of Zuckerkandl or the carotid body. Histologically, neuroblastoma is composed of small round blue cells with high nuclear to cytoplasmic ratio, similar to primitive neuro-ectodermal tumor and Ewing sarcoma. There is a spectrum ranging from high-grade forms of neuroblastoma through an intermediate form of ganglio-neuroblastoma, to the low-grade form of ganglio-neuroma. The differentiation of histological sub-type usually cannot be made based on their imaging appearance. Typically, neuroblastoma presents as a large solid tumor mass which may have extensive areas of hemorrhage and calcification. Spinal extension is common when neuroblastoma originates from the paravertebral sympathetic chain, extending into the spinal canal via the neural foramina and displaying a ‘dumbbell’ shaped appearance. On MRI, neuroblastoma is usually iso-to hyperintense on T1-weighted images and intermediate-to hypo-intense onT2-weighted images, with patchy contrast enhancement. Due to the high nuclear-to-cytoplasmic ratio, these masses demonstrate restricted diffusion. Large masses can cause effacement of the thecal sac and can displace and compress the spinal cord (online bonus image, Figure 19). Detailed discussion of all tumors in this category of spinal tumors is beyond the scope of this manuscript.
Conclusion
A wide variety of neoplasms can involve the pediatric spine. MRI with contrast is the preferred modality for evaluation of spinal cord and spinal canal tumors, while CT scanning is most valuable for imaging osseous spinal neoplasms. Imaging the entire neural axis is recommended for spinal cord tumors, which have a propensity for CSF seeding. Familiarity with imaging characteristics of common pediatric spine tumors is important. Certain features, such as the tumor location and imaging characteristics, along with patient age, can help narrow the differential considerations and, in many cases, permit diagnosis of a specific tumor type. In all cases of spine tumors, information provided by the radiologist regarding the extent of involvement of the spinal cord, spinal canal, and adjacent structures is of utmost value to patient care and management.
References
- Tortori-Donati P, Rossi A, Biancheri R. eds. Pediatric Neuroradiology. Berlin: Springer Verlag; 2005.
- Rossi A, Gandolfo C, Morana G, et al. Tumors of the spine in children. Neuroimaging Clin N Am. 2007;17:17-35.
- Wilson PE, Oleszek JL, Clayton GH. Pediatric spinal cord tumors and masses. J Spinal Cord Med. 2007;30:S15-20.
- Smith AB, Soderlund KA, Rushing EJ, Smirniotopolous JG. Radiologic-pathologic correlation of pediatric and adolescent spinal neoplasms: Part 1, Intramedullary spinal neoplasms. AJR Am J Roentgenol. 2012;198:34-43.
- Koeller KK, Rosenblum RS, Morrison AL. Neoplasms of the spinal cord and filum terminale: Radiologic-pathologic correlation. Radiographics. 2000;20:1721-1749.
- Huisman TA. Pediatric tumors of the spine. Cancer Imaging. 2009;9:S45-48.
- Prayson RA. Disseminated spinal cord astrocytoma with features of gliofibroma: A review of the literature. Clin Neuropathol. 2013;32:298-302.
- Hayden Gephart MG, Lober RM, Arrigo RT, et al. Trends in the diagnosis and treatment of pediatric primary spinal cord tumors. J Neurosurg Pediatr.. 2012;10:555-559.
- Bouffet E, Pierre-Kahn A, Marchal JC, et al., Prognostic factors in pediatric spinal cord astrocytoma. Cancer. 1998;83:2391-2399.
- Hamburger C, Buttner A, Weis S. Ganglioglioma of the spinal cord: Report of two rare cases and review of the literature. Neurosurgery. 1997;41:1410-1415; discussion 14151416.
- Patel U, Pinto RS, Miller DC, et al. MR of spinal cord ganglioglioma. AJNR J Am Neuroradiol. 1998;19:879-887.
- Menashe SJ, Iyer RS. Pediatric spinal neoplasia: A practical imaging overview of intramedullary, intradural, and osseous tumors. Curr Probl Diagn Radiol. 2013;42:249-265.
- Kucia EJ, Maughan PH, Kakarla UK, et al. Surgical technique and outcomes in the treatment of spinal cord ependymomas: part II: myxopapillary ependymoma. Neurosurgery.2011;68:90-94.
- Moeller KK, Coventry S, Jernigan S, Moriarty TM. Atypical teratoid/rhabdoid tumor of the spine. AJNR Am J Neuroradiol. 2007;28:593-595.
- Vougioukas VI, , Gläsker S, Hubbe U, et al. Surgical treatment of hemangioblastomas of the central nervous system in pediatric patients. Childs Nerv Syst. 2006;22:1149-1153.
- Tago M, Terahara A, Shin M, et al., Gamma knife surgery for hemangioblastomas. J Neurosurg. 2005;102 Suppl:171-174.
- Abul-Kasim K, Thurnher MM, McKeever P, Sundgren PC. Intradural spinal tumors: Current classification and MRI features. Neuroradiology. 2008;50:301-314.
- Soderlund KA, Smith AB, Rushing EJ, Smirniotopolous JG. Radiologic-pathologic correlation of pediatric and adolescent spinal neoplasms: Part 2, Intradural extramedullary spinal neoplasms. AJR Am J Roentgenol. 2012;198:44-51.
- Kumar V, Solanki RS. Myxopapillary ependymoma of the filum terminale. Pediatr Radiol. 2009;39:415.
- Koontz NA, Wiens AL, Agarwal A, et al. Schwannomatosis: The overlooked neurofibromatosis? AJR Am J Roentgenol. 2013;200:W646-653.
- Khong PL, Goh WH, Wong VC, et al. MRI of spinal tumors in children with neurofibromatosis 1. AJR Am J Roentgenol. 2003;180:413-417.
- Shaikh MI, Saifuddin A, Pringle J, et al. Spinal osteoblastoma: CT and MRI with pathological correlation. Skeletal Radiol. 1999;28:33-40.
- Azouz EM, Saigal G, Rodriguez MM, Podda A. Langerhans’ cell histiocytosis: Pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005;35:103-115.
- Bartels SA, van Koperen PJ, van der Steeg AF, et al. Presacral masses in children: Presentation, aetiology and risk of malignancy. Colorectal Dis. 2011;13:930-934.
- Shekdar K, Feygin T. Fetal neuroimaging. Neuroimaging Clin N Am. 2011;21:677-703, ix.
- Currarino G, Coln D, Votteler T. Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol. 1981;137: 395-398.
- Moulopoulos LA, Dimopoulos MA. Magnetic resonance imaging of the bone marrow in hematologic malignancies. Blood. 1997;90:2127-2147.
- Liu PI, Ishimaru T, McGregor DH, et al. Autopsy study of granulocytic sarcoma (chloroma) in patients with myelogenous leukemia, Hiroshima-Nagasaki 1949-1969. Cancer. 1973;31:948-955.
- Seok JH, Park J, Kim SK, et al. Granulocytic sarcoma of the spine: MRI and clinical review. AJR Am J Roentgenol. 2010;194:485-489.
- Demir HA, Yalçin B, Büyükpamukçu N, et al. Thoracic neuroblastic tumors in childhood. Pediatr Blood Cancer. 2010;54:885-889.