Deciphering drug-induced interstitial lung disease: A mechanistic approach
Images
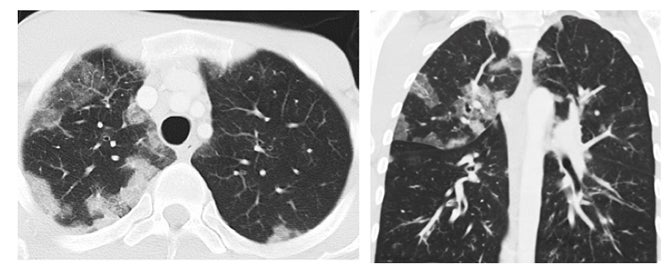
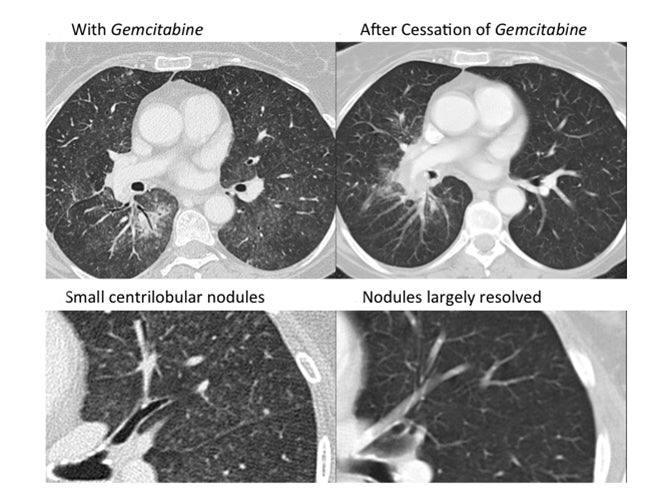
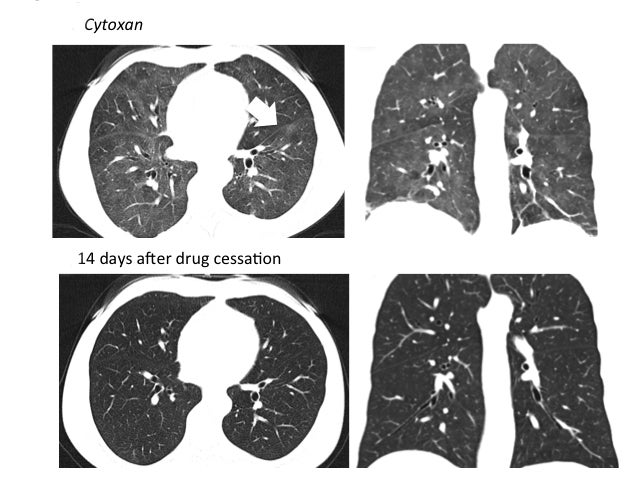
Drug-induced interstitial lung disease (DI-ILD), defined as an interstitial abnormality secondary to administration of a drug, can be acute or chronic.1 Presentation is often insidious and indistinguishable from the underlying disease process being treated by the drug.2 The temporal presentation and correlation are inconsistent and can vary with different drug classes. Due to the continued evolution of drug development in this era of personalized therapy and rapid bench-to-bedside transition of drugs, some of these toxicity patterns may not yet be known. Further, more than 300 medications are reported to cause DI-ILD; the number continues to grow daily.3
In most cases of DI-ILD, patients are relatively asymptomatic and respond well to therapy; therefore, drug-related toxicity often goes unsuspected, especially in its early phases. A high level of suspicion is necessary, however, as most pulmonary drug toxicities are associated with significant morbidity and mortality.2,4 A careful risk-benefit assessment of each drug is warranted, as only routinely successful treatment for toxicity is withdrawal of the offending drug before the injury becomes too extensive or irreversible. Radiologists are often the first to raise a red flag when interpreting images; thus, they are instrumental in modifying patient management in such cases. This article highlights some of the most common causes of drug-induced ILD, as well as some new key offenders, and reviews their presentation.
Overview of ILD
Interstitial lung diseases (ILD) are a heterogeneous group of diseases that produce inflammation and fibrosis of the parenchyma, affecting the alveolar, interstitial and vascular spaces.5 Sometimes the causative etiology or mechanism can be identified (eg, collagen vascular disease, environmental or drug related), while other times it may remain unknown or idiopathic.4
Idiopathic interstitial pneumonias comprise the following disease entities, listed in decreasing order of frequency: Idiopathic pulmonary fibrosis (IPF), nonspecific interstitial pneumonia (NSIP), cryptogenic organizing pneumonia (COP), acute interstitial pneumonia (AIP), respiratory bronchiolitis-associated ILD (RB-ILD), desquamative interstitial pneumonia (DIP) and lymphoid interstitial pneumonia (LIP).6
The first presentation of any ILD is generally shortness of breath or cough.4 Other associated symptoms include fatigue, fever and weight loss. Physical exam findings may include crackles on auscultation, digital clubbing, a restrictive pattern on pulmonary function tests and impaired gas exchange.7 Laboratory studies are mostly non-contributory. A multi-modality approach is often needed to make a diagnosis, with pertinent information and expertise contributed by the pulmonologist, radiologist and pathologist.4 High-resolution CT should be obtained in virtually all patients with a suspected ILD.4 Additional diagnostic workup can include bronchoalveolar lavage, transbronchial biopsy and surgical lung biopsy when indicated.
Drug-induced interstitial lung disease
DI-ILD is a perplexing and challenging diagnosis; in most cases there is no histopathological confirmation and the process is reversible with either removal of the agent or a pulse of steroids after infection is excluded.2,8 Therefore, the diagnosis is based primarily on a radiographic description of interstitial abnormality sub-classified into patterns, with more than one pattern often associated with a particular drug. Various drugs can lead to virtually all histopathological patterns of interstitial pneumonia, depending on the mechanism of injury and healing.1,8 Although certain drug classes tend to cause similar, overlapping patterns of pulmonary involvement,2 others may have more than one presentation depending on severity or mode of drug administration, making early diagnosis even more challenging. The time-to-onset may be quite variable, and can be acute (days to weeks) or subacute (months), often obfuscating the correlation.1
No specific imaging findings distinguish DI-ILD from other causes of ILD. The diagnosis is often made only after other possible causes, such as infection or underlying disease progression, are ruled out.1 One particular challenge is to determine if it is the medication—or the underlying disease it is treating—that is ultimately responsible for pulmonary toxicity.9,10 Early diagnosis and cessation of the causative drug is paramount, as DI-ILD can be fatal.4 Therefore, a high index of suspicion for DI-ILD in any patient on a novel chemotherapeutic regimen with new onset of dyspnea, cough and/or fever is warranted.9
Mechanisms of action of drugs causing ILD
The mechanism of action of toxicity is largely unknown for most drugs and is an active area of research.11 In general, effects can be attributed to direct cell toxicity, free radical production, alterations in collagen production, or indirect inflammatory mediated events.1,2 Understanding these mechanisms can help explain the different imaging patterns of lung toxicity.
Drug-induced diffuse alveolar damage: Pathophysiology and imaging findings
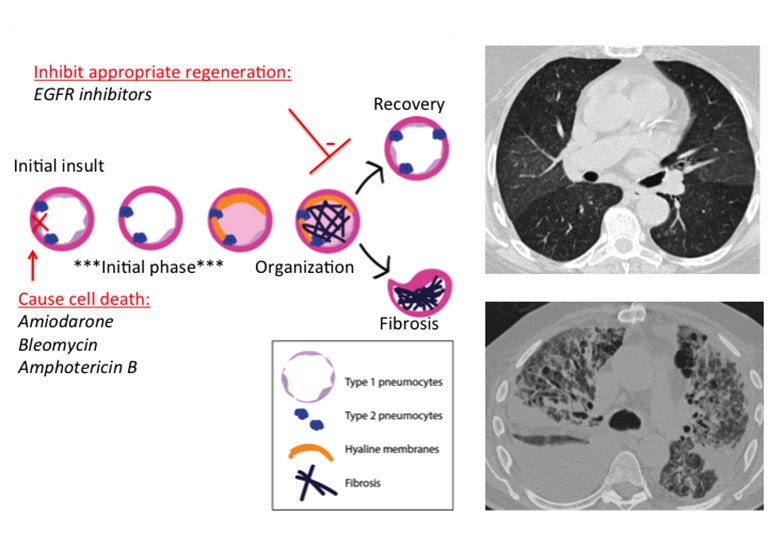
Diffuse alveolar damage (DAD) is a pathology term that describes the presence of alveolar fibrin, hyaline membranes and reactive epithelial cells within alveoli, with varied stages of inflammation.8,12 This is mediated by direct epithelial cell apoptosis and significant damage to the alveolar epithelial barrier (Figure 1).11Acutely, there is cell death of type I pneumocytes, which normally provide a barrier for 97% of the alveolar surface.1 Type II pneumocytes then begin to proliferate and eventually transform into type I pneumocytes to restore the barrier.11 Meanwhile, the alveolar wall is vulnerable and can become edematous; additionally, protein-rich exudates can leak across the weakened wall and flood the alveolus.12 Hyaline membranes--which are effectively a collection of necrotic cell debris and excreted proteins--are secreted to wall off the alveolus and stop oxygen transfer into the damaged alveolus.12 A period of organization then occurs, with organizing fibroblastic tissue filling the alveolar space and interstitium.12 At this point two potential outcomes are possible: recovery and removal of excess fibrin, or permanent fibrosis.
Drugs that disrupt the alveolus via apoptosis or delay the process of barrier restoration could, therefore, lead to DAD and possibly to fibrosis. Bleomycin can cause apoptosis via single- and double-strand DNA breaks and amphotericin B can cause cell-cycle arrest, chromatin condensation and nuclear fragmentation in vitro.13 Amiodarone can cause apoptosis of alveolar epithelial cells and often has a toxicity pattern that includes DAD.14,15 Amiodarone is known to lead to fibrosis, suggesting that it may affect recovery as well (Figure 1). Epidermal growth factor receptors (EGFR) play a role in maintaining and repairing epithelial tissues; thus, EGFR inhibitors could hinder regeneration of the alveolar barrier.11 Interestingly, there are examples in the literature of the EGFR antagonist gefitinib causing diffuse alveolar damage.16,17
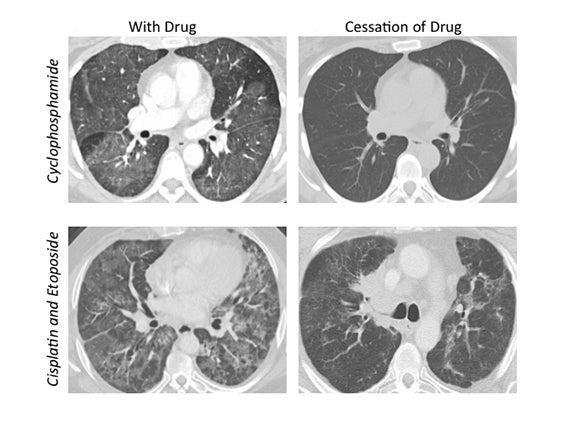
The corresponding imaging findings of DAD include diffuse ground glass opacities and septal thickening with associated areas of consolidative opacities (Figures 1,2).12 In severe cases, consolidative opacities can become confluent.1 There can be significant volume loss in patients who progress to alveolar collapse.12 While DAD can result in fibrosis, it is often reversible.1 Prompt withdrawal of the offending drug is the most effective treatment. Diffuse alveolar damage is currently associated with 51 different drugs on the pneumotox website.3 These include the antiarrhythmic drug amiodarone, the chemotherapeutic agents bleomycin, busulfan, carmustine and cyclophosphamide, the immunosuppressive drugs sirolimus and everolimus and many others.
Indirect drug toxicity via free-radical production and oxidative stress
Indirect cell toxicity from drug metabolites is another pathway by which DI-ILD may occur.2 Of particular importance are drugs that produce reactive oxygen species during metabolism. Oxidative stress may cause apoptosis of structural cells making up the interstitium, thus playing a role in fibrosis.11 This may explain why drugs such as nitrofurantoin and bleomycin are more likely to cause DI-ILD in the presence of supplemental oxygen,8 and can lead to rapid progression of interstitial abnormality on high-flow oxygen therapy.
Immune-mediated drug toxicities: Pathology and imaging findings
Organizing pneumonia
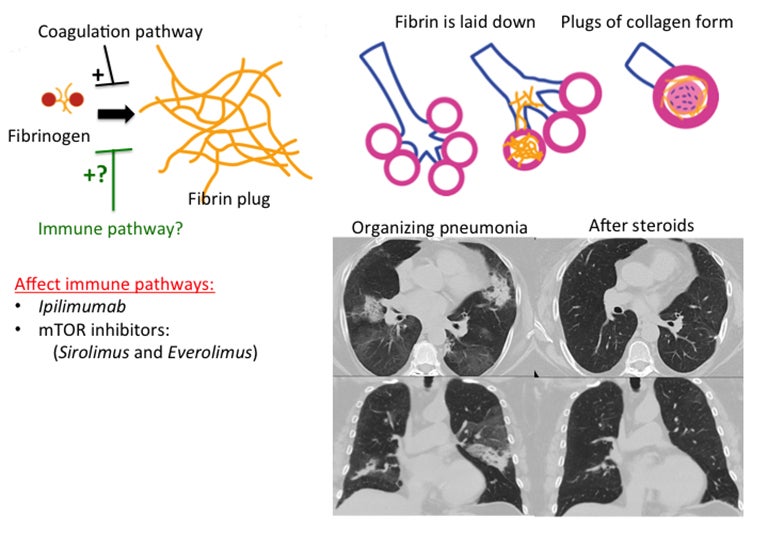
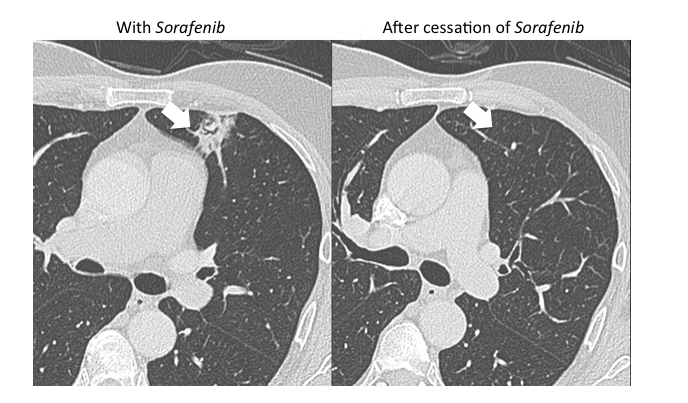
Organizing pneumonia (OP) is characterized by plugs of collagen and fibroblasts that fill respiratory bronchioles, alveolar ducts and alveoli (Figure 3).12 An initial insult leads to fibrin accumulation in the distal airways, and over time this fibrin exudate progresses to bundles of loose collagen that contain spindle-shaped immature fibroblasts (Masson bodies).18 These plugs block aeration of affected distal airways and are effectively the equivalent of a misplaced clot.19 The high lymphocyte count seen in bronchoalveolar lavage (BAL) and the rapid response to steroids suggest that the pathophysiology for organizing pneumonia is immune-related.20 Further, there is at least one report of increased activated T cells in the BAL of patients with OP.21 Organizing pneumonia occurs in patients with a compromised immune system resulting from such conditions as HIV/AIDS and combined variable immunodeficiency syndrome, and in patients who have undergone organ transplantation.22,23 There is increasing evidence that coagulation and innate immune responses may be intertwined, which could explain why an immune insult may lead to the fibrin activation that is a hallmark of OP.19 Various immune-modulating drugs have been described to result in an OP pattern of toxicity. Ipilimumab is a monoclonal antibody that blocks cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and is used to treat melanoma. The drug was shown to cause OP, raising the possibility that its immune-mediating effects were responsible for the observed toxicity pattern.20 mTOR inhibitors such as sirolimus and everolimus are also well-known to cause an organizing pneumonia pattern of drug toxicity.24 mTOR inhibitors inhibit adaptive immunity but enhance the innate immune response.25 Immune-mediated etiology is suggested by the presence of lymphocytes and eosinophils in BAL samples in patients with mTOR inhibitor induced lung toxicity.24 Another proposed mechanism for an immune-mediated toxicity is that the drug binds to plasma proteins and is processed and presented by antigen presenting cells to T lymphocytes, leading to macrophage recruitment.24
The imaging pattern of OP is heterogeneous, but the most common pattern is scattered consolidative opacities in a bilateral and peripheral distribution (Figure 3 and 4).12 Nodular opacities, including centrilobular nodules, can also be observed. A hallmark of this pattern is that opacities tend to migrate on sequential imaging with a waxing and waning appearance.12,18 Other findings that help make the diagnosis are confirmatory pathology, lack of prominent findings on BAL, lack of tissue eosinophilia, and known exposure to the suspected drug. Drugs responsible for this pattern include chemotherapy agents (bleomycin, cyclophosphamide) and immunosuppressive agents (sirolimus, tacrolimus, methotrexate, and gold salts). Steroids and drug cessation are treatment mainstays.
Eosinophilic pneumonia
Eosinophilic pneumonia is similar to organizing pneumonia in pathophysiology, but is mediated instead by an inappropriate increase in the number of eosinophils and macrophages in alveoli. Drugs related to this pathology finding are also often immune modulators. For example, mesalamine, a drug used for ulcerative colitis, has been shown to result in lung toxicity with an eosinophilic pneumonia pattern.26 Daptomycin, implicated in dozens of cases of eosinophilic pneumonia, is thought to act via an antigen-mediated process whereby alveolar macrophages and T-helper cells are activated.27 The imaging hallmark of eosinophilic pneumonia is peripheral opacities with an upper-lobe-predominant distribution (Figure 5). Approximately 115 drugs have been reported to present with this toxicity pattern, including methotrexate and amiodarone. Other drugs responsible for this pattern include immunosuppressive agents (penicillamine), antibiotics (sulfasalazine, nitrofurantoin) and NSAIDS.
Hypersensitivity pneumonitis
Hypersensitivity pneumonitis (HP) is another immune-mediated ILD, usually caused by the inhalation of antigens. Pathologic findings include an accumulation of lymphocytes and plasma cells in the walls of alveoli and bronchioles and cellular bronchiolitis.28,29 Inflammation is followed by the formation of loose nonnecrotizing epitheliod cell granulomas in the bronchiolar wall and alveolar ducts.29 In chronic HP, peribronchiolar and alveolar fibrosis often occurs.30
The term hypersensitivity pneumonitis, when used to describe drug-mediated interactions, may be slightly misplaced, since the mechanism of action in drug-mediated disease is not secondary to inhaled antigens.30 Rather, the term is used to convey the fact that the mechanism of toxicity is believed to be secondary to a drug interaction with the immune system and in cases where the imaging and pathology findings are the same as seen with antigen-mediated HP.31 Like HP caused by inhaled antigens, there is a report of lymphocytosis on BAL, a decrease in CD4 to CD8 ratio and an increase in eosinophils with causative drugs.31
Docetaxal-induced HP has been reported to occur after the 2nd to 4th treatment and is usually mitigated by corticosteroid therapy, raising the possibility that cytotoxic T cells are partly responsible.32 Cephalosporins with identical R1 side chains led to HP in one patient; these R1 side chains can cause hypersensitivity because of the beta-lactam ring structure, but are also implicated in specificity of immune response to cephalosporins.31
The imaging pattern is typically diffuse ground glass opacification without associated interstitial thickening, poorly defined centrilobular ground glass nodules and mosaic attenuation.28 The mosaic attenuation pattern reflects air trapping from underlying obliterative bronchiolitis.28 Chronic HP can progress to an IPF pattern, with reticular opacities and honeycombing.28 Emphysema is another finding seen in chronic HP.28 Hypersensitivity pneumonitis is commonly associated with pencillamine, sulfasalazine, nitrofurantoin, NSAIDS, progesterone, paclitaxel, docetaxel and imatinib.
Pulmonary fibrosis
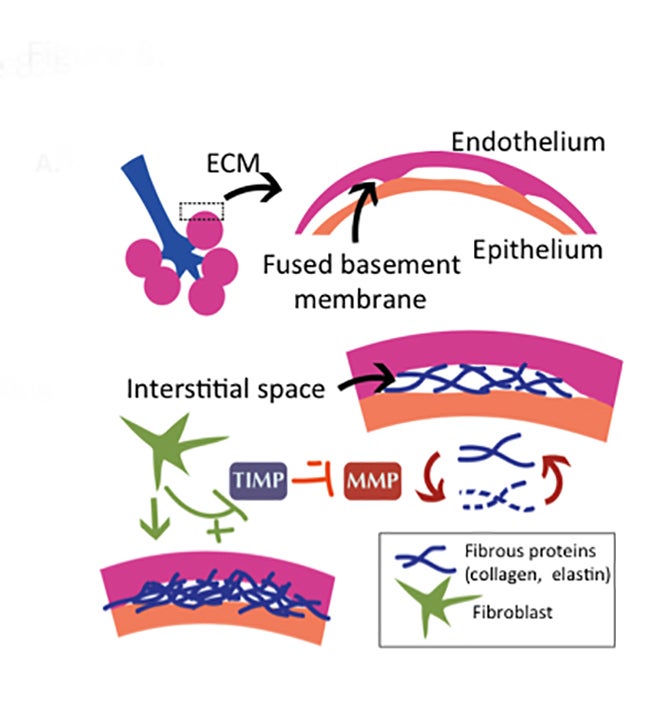
Pulmonary fibrosis is the result of excessive deposition of extracellular matrix proteins in the lung (Figure 8).5 While fibrosis can be a protective response, walling off infections, it can also be pathologic, leading to destruction of normal lung parenchyma and death.5 Fibroblasts normally maintain matrix homeostasis by modulating the turnover of the extracellular membrane (ECM).33 This is orchestrated through the expression of matrix metalloproteinases (MMPs), which degrade the ECM, and expression of tissue inhibitors of metalloproteinases (TIMPs), which block their action and preserve the ECM. In patients with interstitial fibrosis, fibroblasts appear to become abnormally activated and relatively resistant to apoptosis,34 leading, in turn, to over-activation of TIMPs that migrate to sites of injury and promote fibrosis.33 Substantiating this theory, TIMPs are overexpressed in fibrotic lungs.35 Angiogenesis also plays a role in interstitial fibrosis, providing necessary blood supply for the extracellular membrane remodeling.36 Drugs that lead to fibrosis may alter these pathways, favoring the activation of fibroblasts, conferring them with relative resistance to apoptosis or promoting neovascularization that supports ECM remodeling. Bleomycin has been implicated in many of these pathways,37 and recent work has demonstrated that blocking proteins in these pathways can decrease bleomycin-induced fibrosis in animal models.36,38
There are two main imaging patterns of pulmonary fibrosis: Idiopathic pulmonary fibrosis (IPF) and non-specific interstitial pneumonitis (NSIP). NSIP, the most common pattern of fibrosis seen in DI-ILD, is characterized by basal-predominant subpleural reticular opacities, an absence of honeycombing pattern and scattered ground glass opacities.1 An IPF pattern is distinguished from NSIP in that it is associated with honeycombing and traction bronchiectasis and is more likely to be subpleural. Features inconsistent with fibrosis include extensive ground-glass abnormality out of proportion to reticular opacities, micronodules, a mid- or upper lung predominance and consolidative opacities in a bronchopulmonary segment or lobe. There are currently 75 drugs associated with pulmonary fibrosis, including amiodarone, bleomycin, busulfan, carmustine, lomustine and nitrofurantoin.
Pulmonary hemorrhage
Pulmonary hemorrhage is a rare complication of DI-ILD, but is seen with anticoagulants, anti-fungals (amphotericin B), chemotherapeutic agents (mitomycin, Ara-C, high-dose cyclophosphamide), cardiac medications (amiodarone) and immunosuppressive agents (penicillamine).1 Some of these patients will present with hemoptysis in addition to acute dyspnea — penicillamine is particularly implicated in this finding. The radiographic appearance is typically parenchymal opacities in a geographic distribution.
Who is at risk?
Patients react differently to medications, and often genetic and environmental factors can contribute to an atypical reaction.2 Genetic polymorphisms have also been implicated and are a possible explanation of why some groups of people are more likely than others to develop drug toxicity; indeed, the presence of certain variant cytochrome P-450 (CYP) alleles was strongly associated with development of DI-ILD in a Dutch study, with odds ratios ranging from 3.25 to 40.8.39 Dosage is also a factor, particularly with certain medications such as amiodarone, bleomycin and BCNU.2 Extremes of age are also associated with increased risk. Multiple medications will also increase risk, with certain combinations leading to synergistic reaction with worrisome consequences.
Management
Diagnosing DI-ILD remains challenging, as different patterns can overlap and the appearance is often similar to the underlying disease process and infections. The diagnosis is generally made by exclusion and careful linkage of findings and symptoms with administration of the implicated drug.2 In most cases, even after identifying and eliminating the causal agent, transbronchial lavage or biopsy is often needed to exclude infection, prior to administration of steroids. Radiologists play a key role in early identification and triaging the patient to optimal management strategy.
References
- Erasmus JJ, Mcadams HP, Rossi SE. Drug-induced lung injury. Semin Roentgenol. 2002;37(1):72-81.
- Schwaiblmair M, Behr W, Haeckel T, et al. Drug induced interstitial lung disease. Open Respir Med J. 2012;6:63-74.
- Pneumotox online.
- Wallis A, Spinks K. The diagnosis and management of interstitial lung diseases. Br Med J. 2015;350:1-12.
- Mehrad B, Strieter RM. Fibrocytes and the pathogenesis of diffuse parenchymal lung disease. Fibrogenesis Tissue Repair. 2012;5 Suppl 1(Suppl 1):S22.
- Kadoch MA, Cham MD, Beasley MB, et al. Idiopathic interstitial pneumonias: A radiology-pathology correlation based on the revised 2013 American Thoracic Society-European Respiratory Society Classification System. Curr Probl Diagn Radiol. 2015;44(1):15-25.
- American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165(2):277-304.
- Rossi SE, Erasmus JJ, McAdams HP, et al. Pulmonary drug toxicity: Radiologic and pathologic manifestations. Radiographics. 2000;20(5):
1245-1259. - Danson S, Blackhall F, Hulse P, et al. Interstitial lung disease in lung cancer: Separating disease progression from treatment effects. Drug Saf. 2005;28(2):103-113.
- Hallowell RW, Horton MR. Interstitial lung disease in patients with rheumatoid arthritis: spontaneous and drug induced. Drugs. 2014;74(4):443-450.
- Higenbottam T, Kuwano K, Nemery B, et al. Understanding the mechanisms of drug-associated interstitial lung disease. Br J Cancer. 2004;91 Suppl 2:S31-S37.
- Kligerman SJ, Franks TJ, Galvin JR. Organization and fibrosis as a response to lung injury in diffuse alveolar damage, organizing pneumonia and acute fibrinous and organizing pneumonia. Radiographics. 2013;33(7):1951-1975.
- Almeida B, Silva A, Mesquita A, et al. Drug-induced apoptosis in yeast. Biochim Biophys Acta. 2008;1783(7):1436-1448.
- Mahavadi P, Henneke I, Ruppert C, et al. Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone-induced lung fibrosis. Toxicol Sci. 2014;142(1):285-297.
- Bolt MW, Card JW, Racz WJ, et al. Disruption of mitochondrial function and cellular ATP levels by amiodarone and N-desethylamiodarone in initiation of amiodarone-induced pulmonary cytotoxicity. J Pharmacol Exp Ther. 2001;298(3):1280-1289.
- Okamoto I, Fujii K, Matsumoto M, et al. Diffuse alveolar damage after ZD1839 therapy in a patient with non-small cell lung cancer. Lung Cancer. 2003;40(3):339-342.
- Takeda M, Okamoto I, Tsurutani J, et al. Clinical impact of switching to a second EGFR-TKI after a severe AE related to a first EGFR-TKI in EGFR-mutated NSCLC. Jpn J Clin Oncol. 2012;42(6):528-533.
- Cordier JF. Organising pneumonia. Thorax. 2000;55(4):318-328.
- Delvaeye M, Conway EM. Coagulation and innate immune responses: Can we view them separately? Blood. 2009;114(12):2367-2374.
- Barjaktarevic IZ, Qadir N, Suri A, et al. Organizing pneumonia as a side effect of ipilimumab treatment of melanoma. Chest. 2013;143(3):
858-861. - Mukae H, Kadota JI, Kohno S, et al. Increase of activated T-cells in BAL fluid of Japanese patients with bronchiolitis obliterans organizing pneumonia and chronic eosinophilic pneumonia. Chest. 1995;108(1):123-128.
- Khater FJ, Moorman JP, Myers JW, et al. Bronchiolitis obliterans organizing pneumonia as a manifestation of AIDS: Case report and literature review. J Infect. 2004;49(2):159-164.
- Chaparro C, Chamberlain D, Maurer J, et al. Bronchiolitis obliterans organizing pneumonia (BOOP) in lung transplant recipients. Chest. 1996;110(5):1150-1154.
- Baas MC, Struijk GH, Moes DJ, et al. Interstitial pneumonitis caused by everolimus: A case-cohort study in renal transplant recipients. Transpl Int. 2014;27(5):428-436.
- Säemann MD, Haidinger M, Hecking M, et al. The multifunctional role of mTOR in innate immunity: Implications for transplant immunity. Am J Transplant. 2009;9(12):2655-2661.
- Tanigawa K, Sugiyama K, Matsuyama H, et al. Mesalazine-induced eosinophilic pneumonia. Respiration. 1999;66(1):69-72.
- Phillips J, Cardio AP, Patterson TF, et al. Daptomycin-induced acute eosinophilic pneumonia: Analysis of the current data and illustrative case reports. Scand J Infect Dis. 2013;45(10):804-808.
- Hirschmann JV, Pipavath NJ, Godwin, et al. Hypersensitivity pneumonitis: A historical, clinical , and radiologic review. Radiographics. 2009;29(7):1921-1938.
- Baur X, Fischer A, Budnik LT. Spotlight on the diagnosis of extrinsic allergic alveolitis (hypersensitivity pneumonitis). J Occup Med Toxicol. 2015;10(1):15.
- Patel AM, Ryu JH, Reed CE. Hypersensitivity pneumonitis: Current concepts and future questions. J Allergy Clin Immunol. 2001;108(5):661-670.
- Lee SH, Kim M, Lee K, et al. Hypersensitivity pneumonitis caused by cephalosporins with identical R1 side chains. Allergy Asthma Immunol Res. 2014; 25:1-5.
- Taj A. Docetaxel-induced hypersensitivity pneumonitis mimicking lymphangitic carcinomatosis in a patient with metastatic adenocarcinoma of the lung. Hematol Oncol Stem Cell Ther. 2013;6(3-4):117-119.
- Bagnato G, Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. 2015;24(135):102-114.
- Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3(4):350-356.
- Hsu E, Shi H, Jordan RM, et al. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63(3):783-794.
- Iyer AK V, Ramesh V, Castro CA, et al. Nitric oxide mediates bleomycin-induced angiogenesis and pulmonary fibrosis via regulation of VEGF. J Cell Biochem. 2015. doi:10.1002/jcb.25192.
- Blaauboer ME, Boeijen FR, Emson CL, et al. Extracellular matrix proteins: A positive feedback loop in lung fibrosis? Matrix Biol. 2014;34:170-178.
- Wang C, Xuan X, Yao W, et al. Anti-profibrotic effects of artesunate on bleomycin-induced pulmonary fibrosis in Sprague Dawley rats. Mol Med Rep. 2015;12(1):1291-1297.
- Wijnen P, Drent M, Nelemans P, et al. Role of cytochrome P450 polymorphisms in the development of pulmonary drug toxicity: A case-control study in the Netherlands. Drug Saf. 2008;31(12):1125-1134.
- Pelosi P, Rocco PM, Negrini D, et al. The extracellular matrix of the lung and its role in edema formation. An Acad Bras Cienc. 2007;79(2):285-297.
Citation
Ripley, T K, RR G.Deciphering drug-induced interstitial lung disease: A mechanistic approach. Appl Radiol. 2016; (4):9-18.
April 4, 2016