Cystic Fibrosis Liver Disease
Images
CASE SUMMARY
A 13-year-old diagnosed with cystic fibrosis (CF) and pancreatic insufficiency was seen for gastroenterology consultation after being found to have new onset of mild transaminase elevation during an admission for CF pulmonary exacerbation. They showed no jaundice or manifestations of liver disease and denied abdominal complaints, hematemesis, diarrhea, constipation, easy bleeding or bruising, or symptoms of encephalopathy. After an initial work-up, no other causes of liver disease were identified.
IMAGING FINDINGS
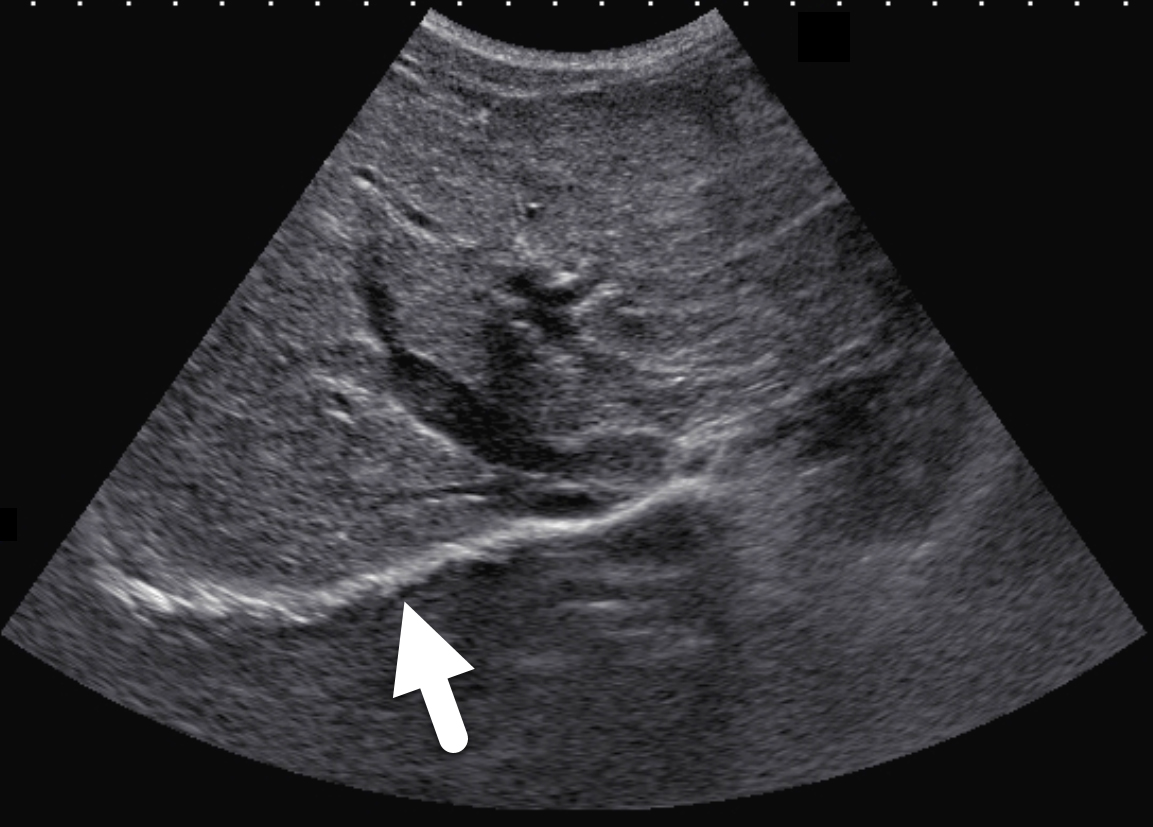
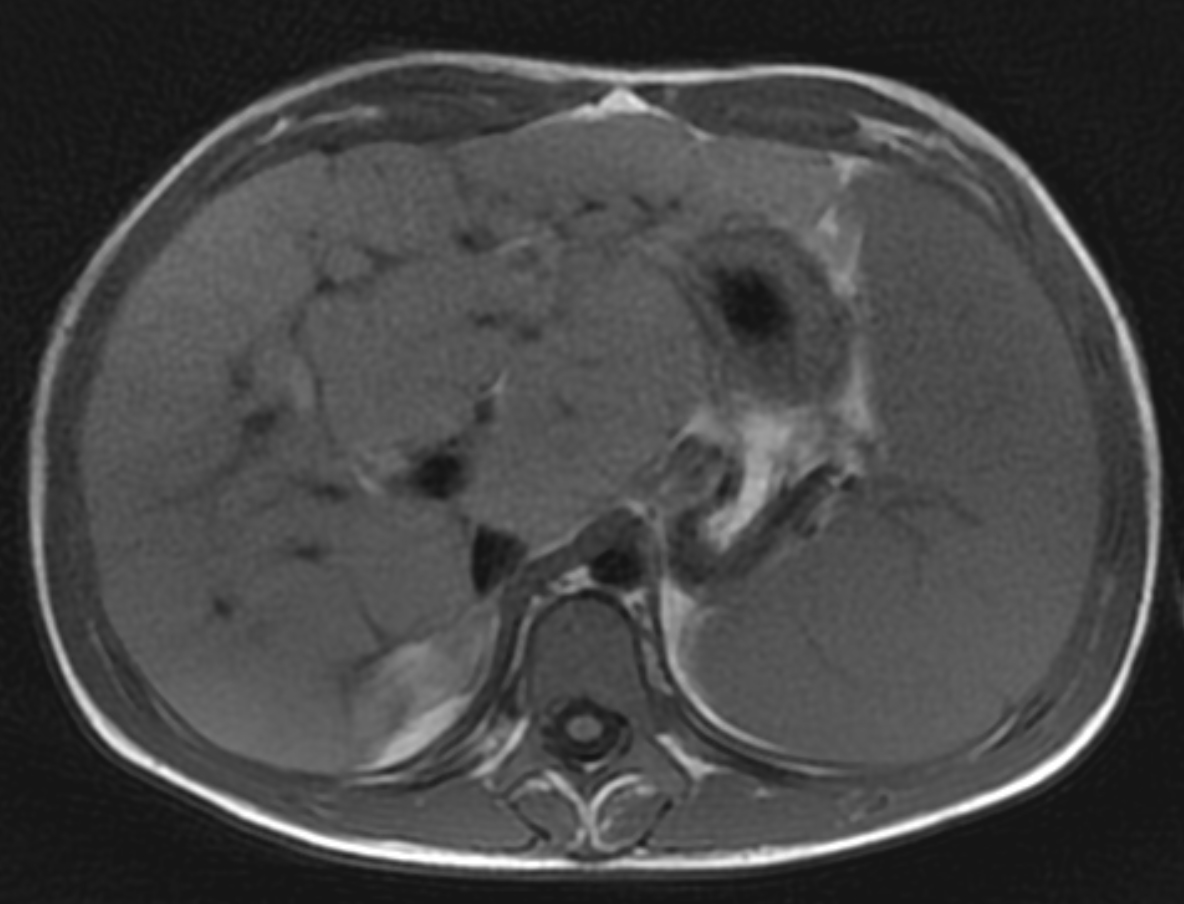
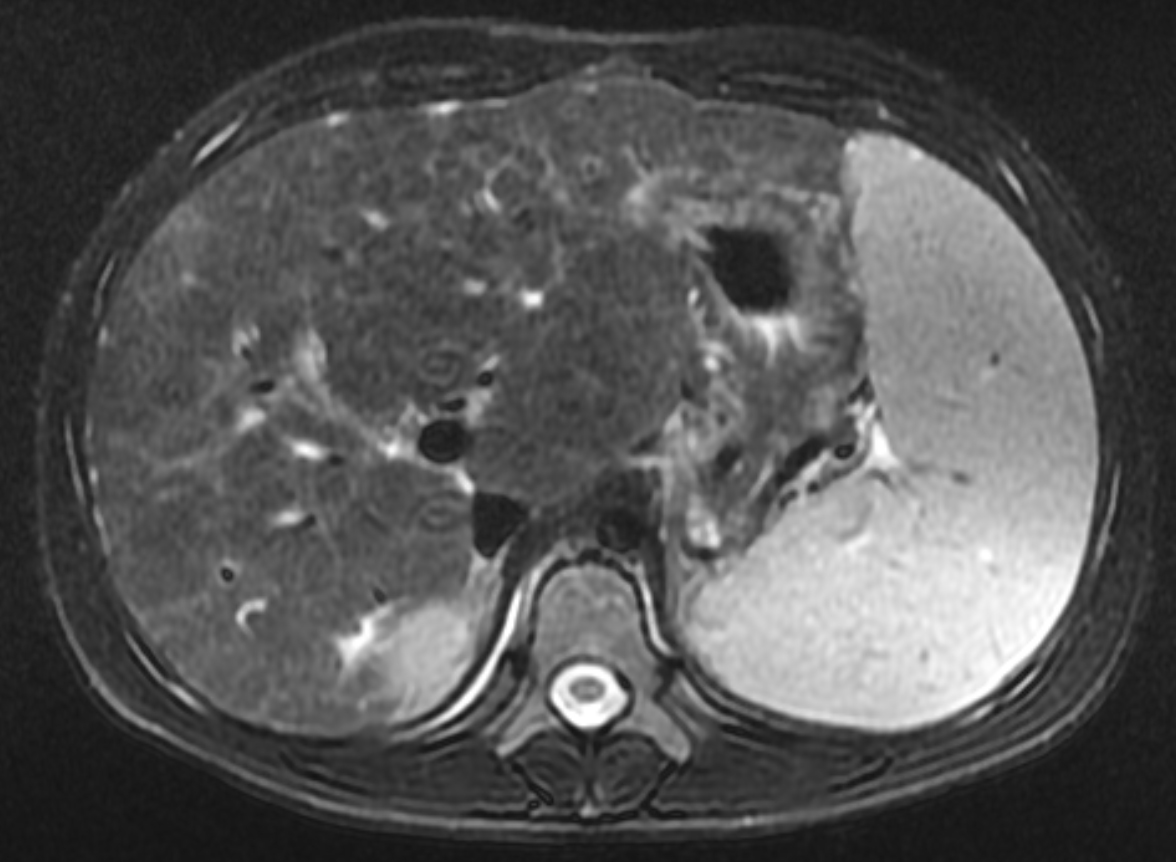
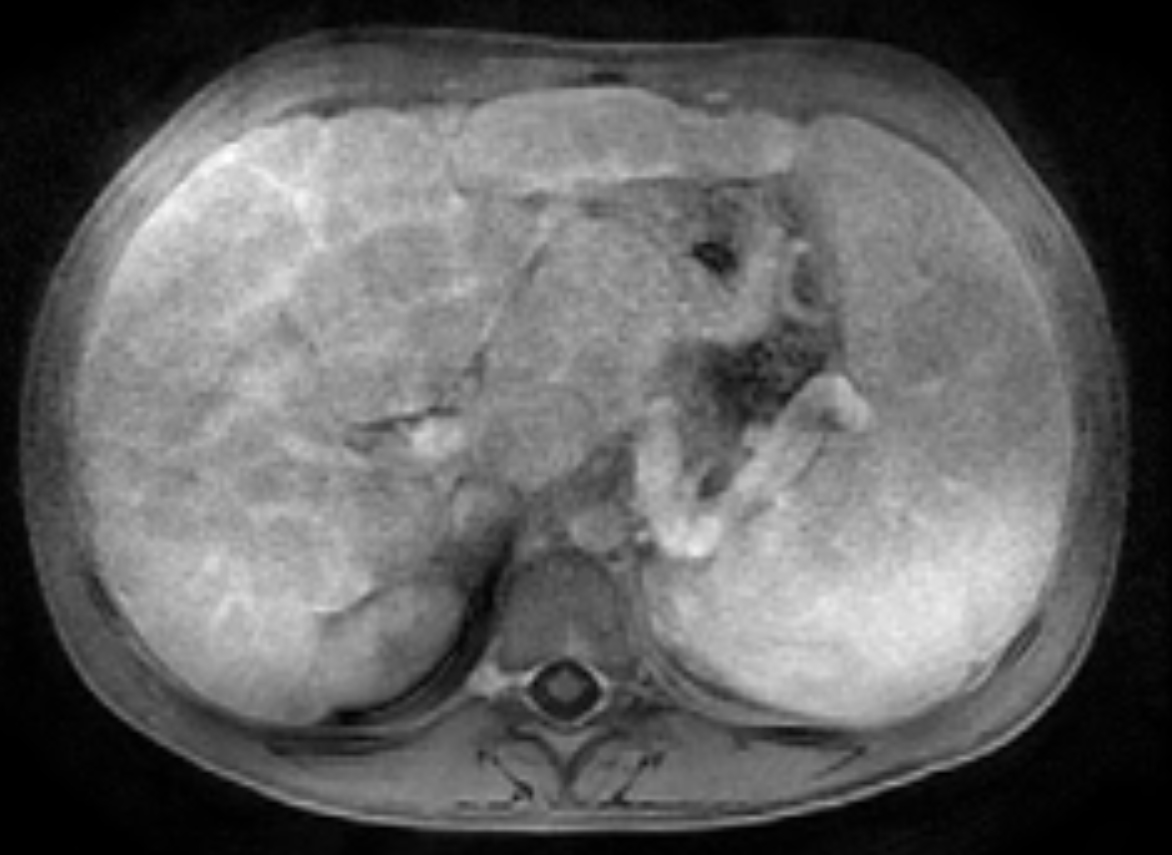
Two years following initial consultation, abdominal ultrasound (Figure 1) showed the liver to have a coarsened, heterogeneous echotexture with a nodular surface. Subsequent MRI (Figure 2), performed 2 years after the initial ultrasound, showed the liver to have a cirrhotic morphology with an irregular, macronodular contour and diffuse periportal edema. There were associated findings of portal hypertension with borderline enlargement of the main portal vein, splenomegaly, and enlarged tortuous splenic vein.
DIAGNOSIS
Cystic fibrosis liver disease (CFLD)
DISCUSSION
Cystic fibrosis is a systemic, autosomal recessive disease that can give rise to complications involving multiple organ systems. As patient life expectancies increase, liver disease has increasingly become recognized as a major consequence of CF. CFLD often arises in children prior to puberty and is believed to be the third-most common cause of death in CF patients.1,2 Reported prevalence rates have ranged from 2 to 37 percent;3 however, owing to its lack of widely agreed-upon diagnostic criteria, its variable imaging and clinical presentations, and slowly progressive nature, CFLD’s true prevalence is unknown.
In the hepatobiliary system, the CF transmembrane conductance regulator (CFTR) protein is expressed in the apical membranes of cholangiocytes and the gallbladder epithelium, where it regulates the water and electrolyte contents and alkalinization of bile. Impaired function of CFTR results in abnormally viscous bile with reduced alkalinity that accumulates in the biliary tree and obstructs the small bile ducts. Abnormal bile composition and reduced flow leads to injury of cholangiocytes and collateral hepatocytes, stimulating the release of inflammatory cytokines and growth factors and inducing hepatic stellate cells to secrete collagen.3 This fibrotic process results in the focal, then multilobular cirrhotic, patterns seen in patients with CFLD. Hepatobiliary complications are believed to occur exclusively in those with severe CFTR gene mutations.2 An important observation that correlates with severe CFTR mutations is that CFLD rarely occurs with pancreatic sufficiency. However, no specific risk factors for liver disease have been identified, and why only certain patients with severe mutations present with CFLD remains unclear.3
CFLD is characterized by three different hepatic parenchymal imaging patterns.3 The first pattern is presumed to represent hepatic steatosis with or without hepatitis. On ultrasound, the liver is enlarged and hyperechoic compared to the right kidney. As fat content increases, the through-transmission of the hepatic parenchyma and visualization of portal triads and the right hemidiaphragm decrease.4 The second imaging pattern is thought to be caused by focal biliary cirrhosis. In this pattern, the liver has a heterogeneous appearance with focal areas of increased periportal echogenicity.4 Finally, the nodular pattern is thought to represent changes related to hepatic fibrosis; it is present in approximately 10% of CFLD patients.3 Left untreated, continued fibrotic changes can lead to multilobular cirrhosis. On ultrasound, the liver appears heterogeneous with a coarsened echotexture and an irregular, nodular margin. As fibrosis progresses, cirrhosis can develop, leading to other complications of hepatic dysfunction, including jaundice, coagulation disorders, portal hypertension, esophageal varices and, as seen in this patient, ascites and splenomegaly.3
Clinically diagnosing CFLD is challenging for two reasons. First, laboratory markers are neither sensitive nor specific for CFLD. While the condition is commonly associated with normal-to-moderate elevations in liver enzymes, these values are similar to the liver enzyme levels found in CF patients without liver disease. Second, most patients are asymptomatic. The most common form of CFLD is hepatic steatosis, which occurs in 20 to 60% of CF patients.4
Ultrasound has been proposed as a screening tool for CFLD. In one study of 719 patients, 18% had abnormal ultrasound liver patterns, with some showing a nodular cirrhotic ultrasound pattern prior to any clinical evidence of liver damage.5 Another study showed a strong correlation between surface nodularity on ultrasound and fibrotic histologic findings on liver biopsy. However, the same study highlighted other patients with histological signs of fibrosis who had normal-appearing ultrasounds. The authors concluded that a negative ultrasound does not preclude diagnosis of CFLD and should be used in concert with other markers of liver disease.6 Based on these findings and the imaging findings previously described, an ultrasound scoring system has been proposed.7 Three points are allotted to findings of liver parenchymal coarseness, liver edge nodularity, and periportal echogenicity, each based on severity, for a potential total of 9 points. Scores of 4 and above correlated with significant elevations of liver enzymes and serum bilirubin and decreased levels of serum albumin and platelet counts.
MRI is also useful in detecting and quantifying liver disease, as it may detect cirrhotic changes with fibrosis, regenerative nodules, portal hypertension, and fatty changes.8 One study identified three MRI findings that reliably distinguished CFLD patients from a control group. These were altered gallbladder morphology, periportal tracking on diffusion weighted imaging, and periportal fat deposition in chemical-shift imaging.9 More recent MR imaging techniques allow for quantification of the different patterns of liver disease. Objectives measures such as liver stiffness, liver fat fraction, liver volume and spleen volume can be used to quantitatively evaluate the liver.
Currently, ursodeoxycholic acid (UDCA) is the main pharmacologic treatment for CFLD. Originally used to treat gallstones, UDCA is believed to improve bile acid flow. However, there are few longitudinal randomized controlled trials studying UDCA therapy for CFLD. In a systematic review of randomized controlled trials comparing the efficacy of UDCA to placebo, there was no difference in rate of portal hypertension or improvement in abnormal biliary excretion.10 Treatment also includes nutritional therapy with optimizing caloric intake, pancreatic enzyme replacement, and fat-soluble vitamin supplementation. For patients with severe portal hypertension, a surgical portosystemic shunt may be used to reduce the risk of life-threatening variceal bleeding. As with other decompensated liver diseases, the only curative treatment for advanced CFLD is liver transplant.
Our patient has remained asymptomatic despite evidence of more advanced liver disease.
CONCLUSION
Hepatobiliary complications have become increasingly relevant in CF as treatments and patient life expectancy continue to improve. Hepatic US and MRI remain the least-invasive methods of CFLD screening. However, CFLD remains difficult to diagnose, as clinical signs often manifest well after liver disease has advanced. As this disease arises largely in the pediatric population, the need remains for improved screening measures. Medical management is currently limited to proper nutrition, fat-soluble vitamin supplementation, and UDCA, although there is little evidence for its efficacy.
REFERENCES
- Stonebraker JR, Ooi CY, Pace RG, et al. Features of Severe Liver Disease with Portal Hypertension in Patients With Cystic Fibrosis. Clinical Gastroenterology and Hepatology. 2016;14(8). doi:10.1016/j.cgh.2016.03.041.
- Colombo C. Liver disease in cystic fibrosis: A prospective study on incidence, risk factors, and outcome. Hepatology. 2002;36(6):1374-1382. doi:10.1053/jhep.2002.37136.
- Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Gastroenterology Review. 2014; 3:136-141. doi:10.5114/pg.2014.43574.
- Diwakar V, Pearson L, Beath S. Liver disease in children with cystic fibrosis. Paediatric Respiratory Reviews. 2001;2(4):340-349. doi:10.1053/prrv.2001.0170.
- Leung DH, Ye W, Molleston JP, Weymann A, Ling S, Paranjape SM, Romero R, Schwarzenberg SJ, Palermo J, Alonso EM, et al. Baseline ultrasound and clinical correlates in children with cystic fibrosis. J Pediatr. 2015; 167:862–868.e2.
- Mueller-Abt PR, Frawley KJ, Greer RM, Lewindon PJ. Comparison of ultrasound and biopsy findings in children with cystic fibrosis related liver disease. J Cystic Fibrosis. 2008;7(3):215-221. doi: 10.1016/j.jcf.2007.08.001.
- Williams SGJ, Evanson JE, Barrett N, Hodson ME, Boultbee JE, Westaby D. An ultrasound scoring system for the diagnosis of liver disease in cystic fibrosis. J Hepatol. 1995; 22:513-521.
- King LJ, Scurr ED, Murugan N, Williams SGJ, Westaby D, Healy JC. Hepatobiliary and Pancreatic Manifestations of Cystic Fibrosis: MR Imaging Appearances. RadioGraphics. 2000;20(3):767-777. doi:10.1148/radiographics.20.3.g00ma08767.
- Poetter-Lang S, Staufer K, Baltzer P, et al. The Efficacy of MRI in the diagnostic workup of cystic fibrosis-associated liver disease: A clinical observational cohort study. Euro Radiol. 2018;29(2):1048-1058. doi:10.1007/s00330-018-5650-5.
- Cheng K, Ashby D, Smyth RL. Ursodeoxycholic acid for cystic fibrosis–related liver disease. Cochrane Database Syst Rev 2017;9:CD000222.
Citation
D C, JJ P, R T, AJ T.Cystic Fibrosis Liver Disease . Appl Radiol. 2020; (5):44-46.
September 1, 2020