Askin tumor
Images

CASE SUMMARY
A previously healthy 9-year-old male presented with a several-week history of progressive shortness of breath.
IMAGING FINDINGS
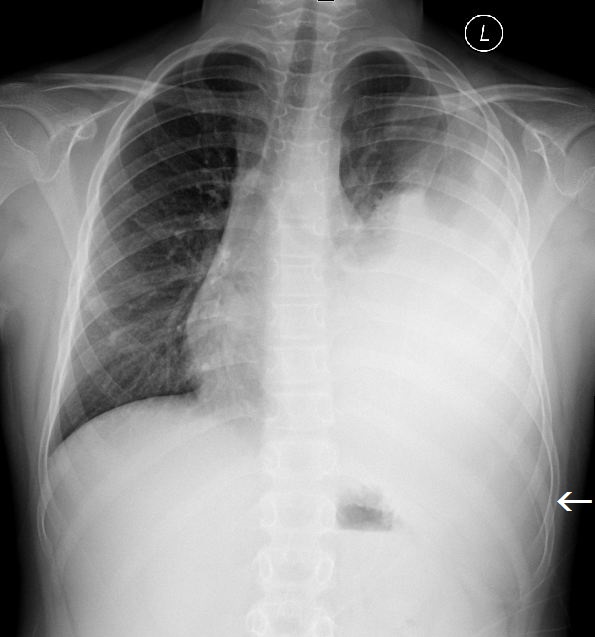
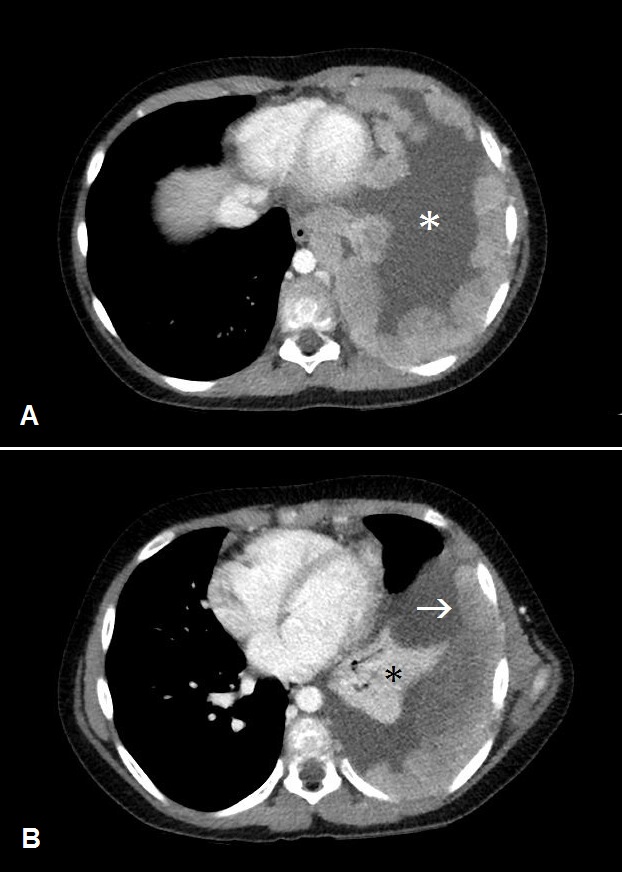
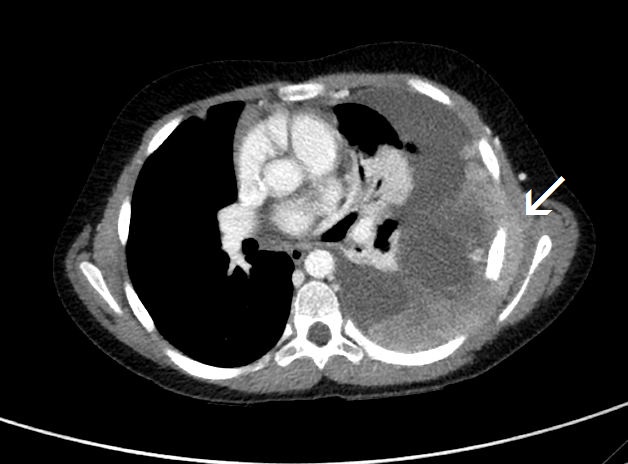
The initial chest radiograph showed a large, poorly defined mass occupying the left hemithorax (Figure 1). There was no splaying or erosion of the ribs. Subsequent chest CT with IV contrast showed pleural-based conglomerate lobular masses of the left hemithorax (Figure 2). The pleural involvement was accompanied by extrapleural extension with encasement of the left seventh rib (Figure 3). Large, left pleural effusion was present along with left lung atelectasis. The CT images in the lung window showed contralateral right lung metastatic lesions (Figure 4).
DIAGNOSIS
Askin tumor (Ewing sarcoma family of tumors).
Differential diagnosis includes other sarcomas such as chondrosarcoma, pleuropulmonary blastoma, lymphoma, and infection.
DISCUSSION
Frederic Askin, MD, and his colleagues initially described a series of small, round, blue cell tumors originating in the soft tissues of the chest wall.1 While the tumor was initially termed a primitive neuroectodermal tumor (PNET), it is now recognized as a distinct entity residing in a family of aggressive types of Ewing sarcoma. Other entities that make up the Ewing sarcoma family of tumors include Ewing sarcoma, peripheral primitive neuroectodermal tumor (pPNET), and neuropithelioma.2 Although the literature is littered with contradictory terminology, these tumors are likely to represent the same entity with minor differences in differentiation. For example, PNET is more differentiated in its expression of neural elements. The Ewing sarcoma family of tumors are similar in that they demonstrate non-random t(11;22)(q24;q12) chromosome rearrangements resulting in the formation of the Ewing sarcoma E26 transformation-specific fusion gene.2
Patients with an Askin tumor typically present in the second or third decade of life, with pain localizing to the chest, back or shoulder.2 Other symptoms include shortness of breath, weight loss and cough. While there is no sex predilection for Askin tumor, patients are nine times more likely to be Caucasian.2
Imaging findings include a large extrapulmonary mass with chest wall extension.3 The mass may be solitary or multiple with an eccentric growth pattern along the chest wall. Often, the exact site of origin is difficult to determine. Rib destruction is present in 25-63% of patients.4 Pleural effusions, lymphadenopathy, and both lung and bone metastases are commonly seen.5,6
Treatment and prognosis vary widely depending on tumor size and metastases. Localized small tumors without metastases can be surgically treated with addition of chemotherapy and/or radiation.3 Bulky tumors and patients with metastatic disease are treated with radiation and chemotherapy. In these patients surgical excision may also be required based on tumor size, location and its associated symptoms. The overall survival for patients with Askin tumor is less than 40% at 2 years.3
CONCLUSION
Askin tumors are a part of the Ewing sarcoma family of tumors. While histologically the tumors are similar to a Ewing sarcoma, their typical location and growth pattern make the tumor a distinct radiographic entity.
REFERENCES
- Tateishi U, Gladish G, Kusumoto M et al. Chest wall tumors: Radiologic findings and pathologic correlation: Part 2. Malignant tumors. Radiographics. 2003; 23:1491-1508.
- Burchill S Ewing’s sarcoma: Diagnostic, prognostic, and therapeutic implications of molecular abnormalities. J Clin Pathol. 2003; 56:96-102.
- Gladish G, Sabloff B, Munden R et al. Primary thoracic sarcomas. Radiographics. 2002: 22:621-637.
- Gurney J, Winer-Muram H, Stern E. (2006) Diagnostic imaging: Chest, 1st edn. Amirsys Publishing, Salt Lake City, Utah.
- Schulman H, Newman-Heinman N, Kurtzbart E et al. Thoracoabdominal peripheral primitive neuroectodermal tumors in childhood: Radiological features. Eur Radiol. 2000;10:1649-1652.
- Sallustio G, Pirronti T, Lasorella A et al. Diagnostic imaging of primitive neuroectodermal tumour of the chest wall (Askin tumour). Pediatr Radiol. 1998; 28:697-702.
Citation
B K, SA J, AJ T, R T.Askin tumor. Appl Radiol. 2017; (6):32-33.
June 8, 2017