Acute presentation of Lhermitte-Duclos disease in an adult patient in association with Cowden syndrome
Images


CASE SUMMARY
A 33-year-old female presented to the emergency department with the chief complaint of new onset severe headache, persisting for five days, with accompanying subjective intermittent ataxia. There was no relevant history of prior migraines or similar such sentinel events. Review of systems was noncontributory. CT imaging demonstrated a nonspecific hypodense mass centered in the superior right cerebellum with mild locoregional mass effect upon the fourth ventricle without evidence of obstructive hydrocephalus. The appearance was nonspecific and considerations included subacute infarct, infiltrating glioma or cerebellitis. Subsequently, MRI was performed, demonstrating the characteristic striated morphologic appearance of dysplastic gangliocytoma or so-called Lhermitte-Duclos disease. Given the prior relevant surgical history of thyroidectomy for goiter and subcutaneous lipoma resection, the findings were felt to be in conjunction with previously undiagnosed Cowden syndrome. The patient was discharged after symptomatic pain control and scheduled non-operative follow up with neurosurgery as an outpatient. The relationship with Cowden syndrome is both clinically relevant and pertinent as well due to the heightened predisposition in these patients for a vicissitude of both benign and malignant neoplasms involving the breast, thyroid, dermis, gastrointestinal, genitourinary and central nervous systems, with annual surveillance and screening measures being highly warranted.
Lhermitte-Duclos disease, so named for French neurologists Jacques Lhermitte and P Duclos after their initial description of the entity in 1920, has undergone a spectrum of growth in pathophysiologic understanding recently and is now considered to be a dysplastic gangliocytoma of the cerebellum, a WHO grade 1 tumor.1 Growth is slow and there is no reported malignant potential with clinical presentation typically entailing signs of increased intracranial pressure, obstructive hydrocephalus, and cerebellar dysfunction.1,2 The MR imaging appearance is considered diagnostic and can obviate biopsy, consisting of a tigroid, striated appearance of the nonenhancing cerebellar mass with superficial bands of alternating T2 hyper- and isointensity to grey matter, iso- and hypointense on T1-weighted imaging, with sporadic calcification reported and minimal loco-regional mass effect.3,4 A strong association with Cowden syndrome, an autosomal dominant, multigenic group of germ line mutations, is now recognized. Due to the heightened risk for a host of potential neuroectodermal related malignancies associated with this phakomatosis, patients are advised to undergo appropriate targeted annual screening and surveillance. As such, the radiologist may play an important role in first suggesting the underlying syndrome based on initial neuroimaging findings.
IMAGING FINDINGS
Our patient, a 33-year-old female, presented to the ER with a five-day history of severe headache and subjective dizziness and ataxia. There was no prior history of prodromal symptoms or migraines and review of systems as well as laboratory data were noncontributory.
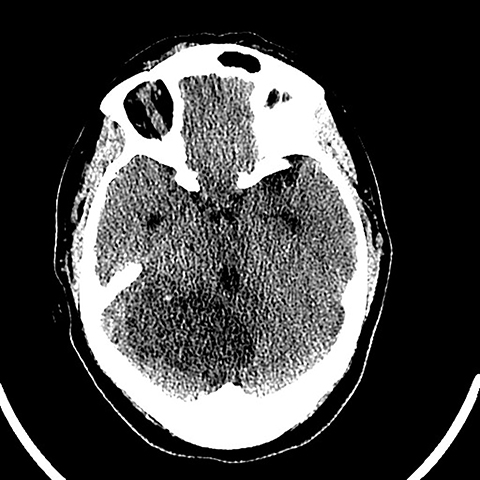
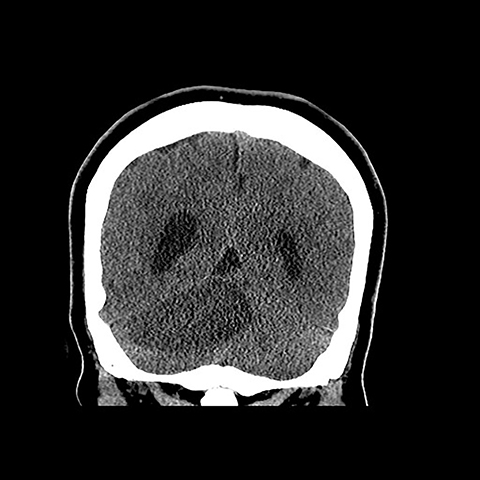
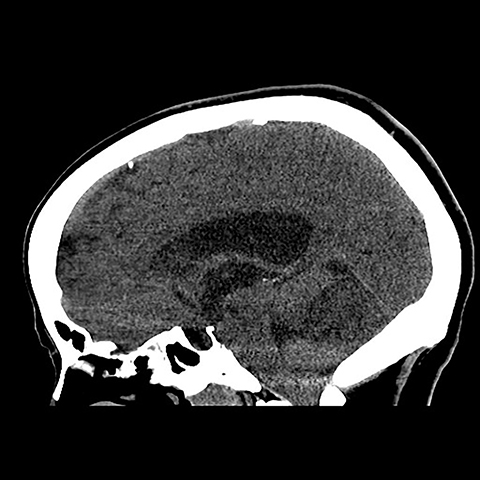
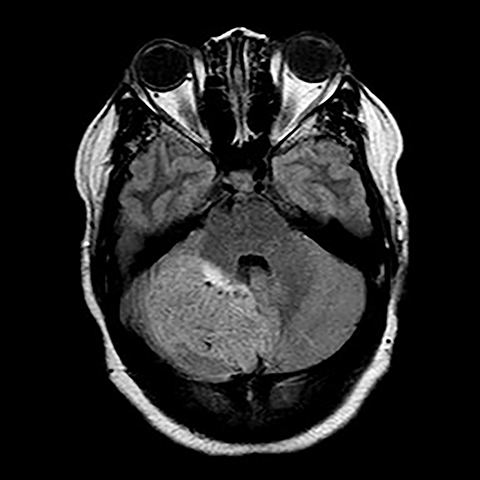
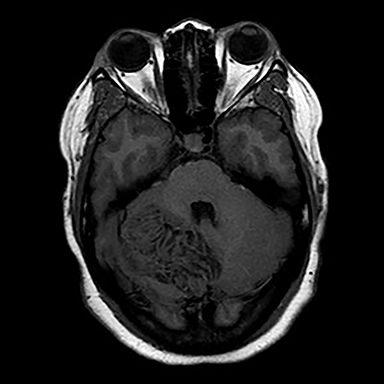
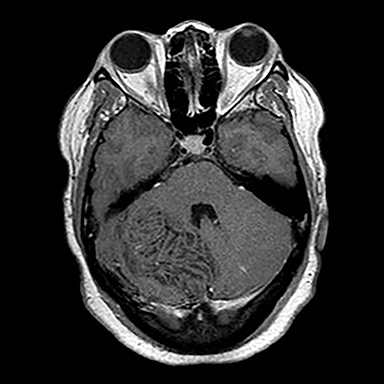
Surgical history was notable for a prior thyroidectomy for benign goiter and previous subcutaneous lipoma resection. Noncontrast CT imaging of the head performed in the ER demonstrated a roughly 6 cm mass centered in the superior right cerebellar hemisphere with cortical involvement and mild mass effect upon the fourth ventricle. A solitary punctate superficial focus of hyperdensity was seen, deemed equivocal for focal hemorrhage or dystrophic calcification. No obstructive hydrocephalus was observed (Figures 1-3). Diagnostic considerations included glioma, subacute infarct or, less likely, cerebellitis. MRI was ordered for further elucidation and proved definitive, demonstrating a focal mass centered in the superior right cerebellum with pathognomonic alternating bands of hypo and hyperintensity on T1 and T2 weighted imaging without restricted diffusion or enhancement. There was minimal surrounding edema and mild mass effect upon the fourth ventricle without evidence of obstructive hydrocephalus (Figures 4-8). The findings were deemed consistent with Lhermitte-Duclos disease. Because of the relevant surgical history and imaging appearance, suspicion for underlying Cowden syndrome was raised. The patient was treated for symptomatic control of her headache, and, after multidisciplinary discussion, was discharged with follow up as an outpatient in the neurosurgical service for surgical planning and clinical surveillance.
DIAGNOSIS
Acute presentation of Lhermitte-Duclos disease in an adult patient in association with Cowden Syndrome
DISCUSSION
Acute presentation of Lhermitte-Duclos disease, or benign dysplastic cerebellar gangliocytoma, in an adult patient is a rather uncommonly described entity in the literature.1,2,4 There has been considerable and ongoing debate as to the correct pathophysiologic classification as a hamartomatous lesion or true neoplasm. Currently it is deemed a World Health Organization grade one tumor, benign, but slowly progressive with potential for recurrence. Histopathogical findings include widening of the molecular layer of the cerebellar cortex with replacement by abnormal ganglion cells, absence of the Purkinje cell layer, and hypertrophy of the granular cell layer.5 Clinically, the duration of symptoms is variable, ranging from a few months to over 10 years. Presentation usually entails signs and symptoms of cerebellar dysfunction and raised intracranial pressure. Visual disturbances, cranial nerve palsies, and focal sensory and motor deficits have all been described.5
Imaging features are pathognomonic, consisting of the well-described tigroid or folial appearance of alternating superficial bands of hyper- and hypointensity on T1 and T2 weighted imaging within the cerebellar lesion. Enhancement is not typically seen nor is there restricted diffusion though T2 shine through effects can be confounding without reference to the ADC maps.3 Spectroscopic analysis demonstrates nonspecific tumoral like findings of decreased NAA and increased lactate, though without the inceased choline to creatine ratio and myo-inositol levels as seen in glial malignancies.3 Regional increased blood cerebral blood volume and flow can be seen on perfusion weighted imaging, thought to correlate with the histological findings of proliferative thin walled and dilated vessels seen on pathologic sections.3 Differential imaging considerations, including hemangioblastoma, glial tumor, primitive neuroectodermal tumor , subacute infarct, cerebellitis, or tumefactive demyelination are limited by the pathognomonic appearance on MRI, though recently there have been several case reports of infiltrating medulloblastoma mimicking Lhermitte-Duclos disease in both pediatric and adult patients. Grossly, in these instances, a lamellated superficial appearance was anecdotally observed on MR imaging, though without the typical hyperdensity of most medulloblastomas on CT secondary to dense hypercellularity. Also lacking were the usual elevated Cho/Cr ratio on spectroscopy or restricted diffusion seen with PNET tumors.6,7
Recent efforts have focused on the relationship of Lhermitte-Duclos disease with Cowden syndrome, a phakomatosis characterized by a multigenic autosomal dominant inheritance pattern and manifesting with a variety of neuroectodermal derived neoplasms, both benign and malignant. According to the International Cowden Consortium Criteria, clinical findings are subdivided into pathognomonic, major, and minor criteria for diagnosis, with major findings including breast, thyroid or endometrial carcinoma, or macrocephaly. Minor criteria include other non-malignant thyroid or breast lesions, fibrolipomatous lesions, gastrointestinal hamartomas, mental retardation or genitourinary tumors. So-called pathognomonic criteria include Lhermitte-Duclos disease and characteristic mucocutaneous lesions such as facial trichlemmomas, acral keratosis, and papillomatous pustules. Clinical diagnosis entails either mucocutaneous lesions, when present to a specified extent, two major criteria - one of which must be Lhermitte Duclos disease or macrocephaly, one major and three minor, or four minor findings.8
Genetically, Cowden syndrome is one of a group of clinical disorders occurring in 1 in 200,000 individuals that have been linked to germline mutations of the phosphatase and tensin homolog PTEN tumor suppressor gene on chromosome 10, and also include Banayan-Riley-Ruvalacaba and Proteus syndromes as well as the more vaguely described group of autism with macrocephaly disorders.8,9 Approximately 80 percent of patients with a clinical diagnosis of Cowden syndrome will have a detectable germline coding sequence mutation in PTEN.8 Among those with the mutation, phenotypic expression is seen in over 99% of individuals by age 30.1 Because of increased risk of cancer, most commonly breast, thyroid or endometrium, Cowden syndrome patients are advised to undergo lifelong surveillance. Average estimated life time risk for breast cancer is 25-50%, 10% for thyroid cancer, and 5-10% for endometrial cancer.10 Surveillance should include annual physical examinations beginning at 18 years of age, annual urinalysis and PAP smears, baseline colonoscopy at age 50, annual mammography, breast MRI and clinical breast exam beginning at age 30-35, baseline thyroid ultrasound at age 18 with annual clinical exam, annual endometrial suction biopsies beginning at 35-40 years of age and annual transvaginal ultrasound in post-menopausal women.9,10 An increased incidence of CNS vascular anomalies, including venous and cavernous angiomas and arteriovenous malformations have also been observed in Cowden patients though on an anecdotal scale and felt not to warrant dedicated screening measures at this time.9
CONCLUSION
In summary, Lhermitte-Duclos disease, or dysplastic gangliocytoma of the cerebellum, is a pathognomonic radiologic entity, the diagnosis of which should remain not only in the bailiwick of neuroimaging specialists but general radiologists as well. Recognition of the underlying relationship with Cowden syndrome is paramount for both genetic counseling and timely patient induction into an appropriate clinical surveillance regimen.
REFERENCES
- Hariri O, Khachekian A, Muilli D, Amin J, Minassian T, Berman B, Ritter Y, Siddiqi J. Acute-onset cerebellar symptoms in Lhermitte-Duclos disease: case report. Cerebellum. 2013;12:127-130.
- Yagci-Kupeli Y, Oguz K, Bilen M, Yalcin B, Akalan N, Buyukpamukcu M. An unusual cause of posterior fossa mass: Lhermitte-Duclos disease. J of the Neurological Sciences. 2010;290:138-141.
- Klisch J, Juengling F, Spreer J, Koch D, Thiel T, Buchert M, Arnold S, Feuerhake F, Schumacher M. Lhermitte-Duclos disease: Assessment with MR imaging, positron emission tomography, single-photon emission CT, and MR spectroscopy. AJNR. 2001;22:824-830.
- Shinagare A, Patil N, Sorte S. Case 144: Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease). Radiology. 2009;251:298-303.
- Nowak D, Trost H. Lhermitte-Duclos disease (dysplastic cerebellar gangliocytoma): a malformation, hamartoma or neoplasm? Acta Neurol Scand. 2002;105:137-145.
- Kamble R, Mathew S, Rao R. Infiltrating medulloblastoma in a child mimicking Lhermitte-Duclos disease. J Pediatr Neurosci. 2012;7(2):159-160.
- Douglas-Akinwande A, Payner T, Hattab E. Medulloblastoma mimicking Lhermitte-Duclos disease on MRI and CT. Clin Neurol and Neurosurg. 2009;111:536-539.
- Pilarski R, Stephens J, Noss R, Fisher J, Prior T. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genetics. 2013;48:505-512.
- Pilarski R. Cowden syndrome: A critical review of the clinical literature. J Genetic Counsel. 2009;18: 13-27.
- Tutluer S, Tanriover M, Guven G. Cowden syndrome: a major indication for extensive cancer surveillance. Med Oncol. 2012;29:1365-1368.
Citation
Rheinboldt, M, Z D, B BH.Acute presentation of Lhermitte-Duclos disease in an adult patient in association with Cowden syndrome. Appl Radiol. 2016; (8):28-31.
August 1, 2016