Surfactant Protein C Deficiency-associated Diffuse Lung Disease
Images
Case Summary
A teenager with known surfactant protein C deficiency and restrictive lung disease, who was extremely sick as an infant, presented with shortness of breath, exercise intolerance, chest pain, palpitations, and severe pectus excavatum (PE) deformity. Echocardiography showed external compression of the right atrium and right ventricle that was hemodynamically insignificant. Flexible bronchoscopy revealed pulsatile, 40-50% extrinsic compression of the left mainstem bronchus anteriorly. They had undergone an extensive workup for hypoxemia and failure to thrive as an infant, including a lung biopsy at four months of age that showed findings of chronic pneumonitis of infancy. Genetic testing revealed the diagnosis of Surfactant Protein C (SP-C) deficiency.
Imaging Findings
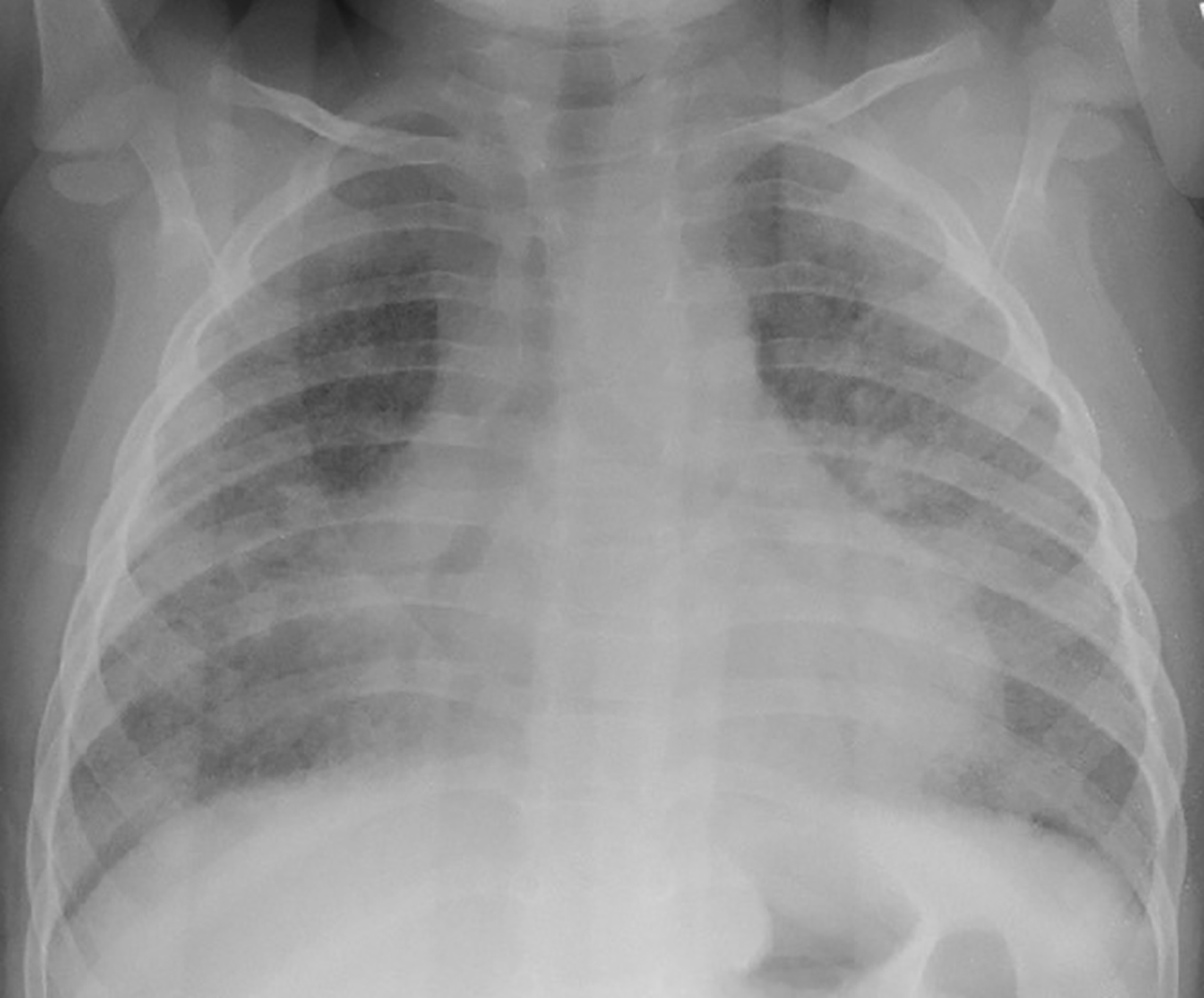
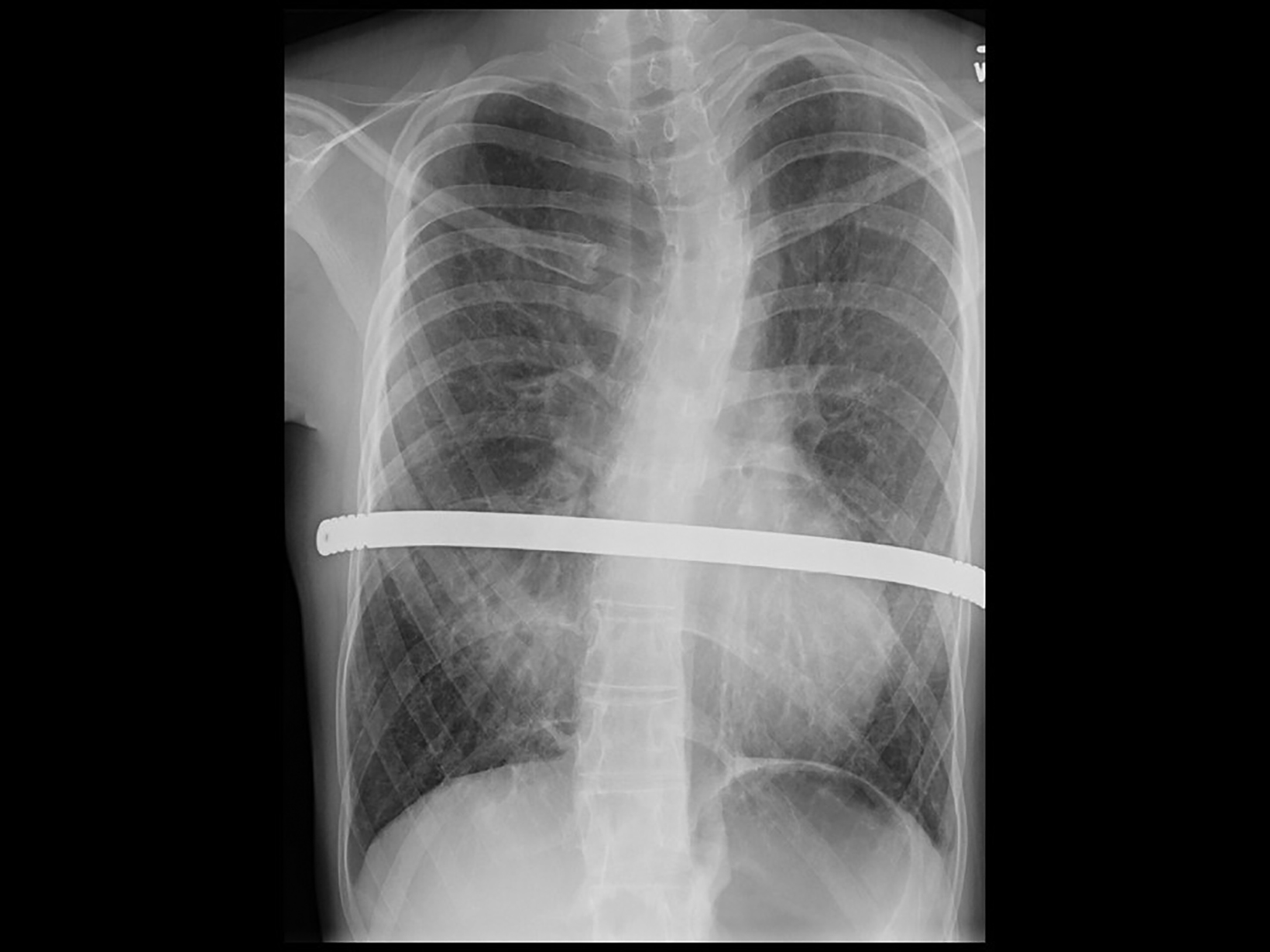
Neonatal chest radiographs showed diffuse haziness in both lung fields. The lungs demonstrated diffuse prominent interstitial markings on the chest radiographs obtained as a toddler with follow-up radiographs showing progressive interstitial lung disease in a bilateral interstitial reticulonodular pattern (Figure 1). With age, the interstitial lung markings became coarser.
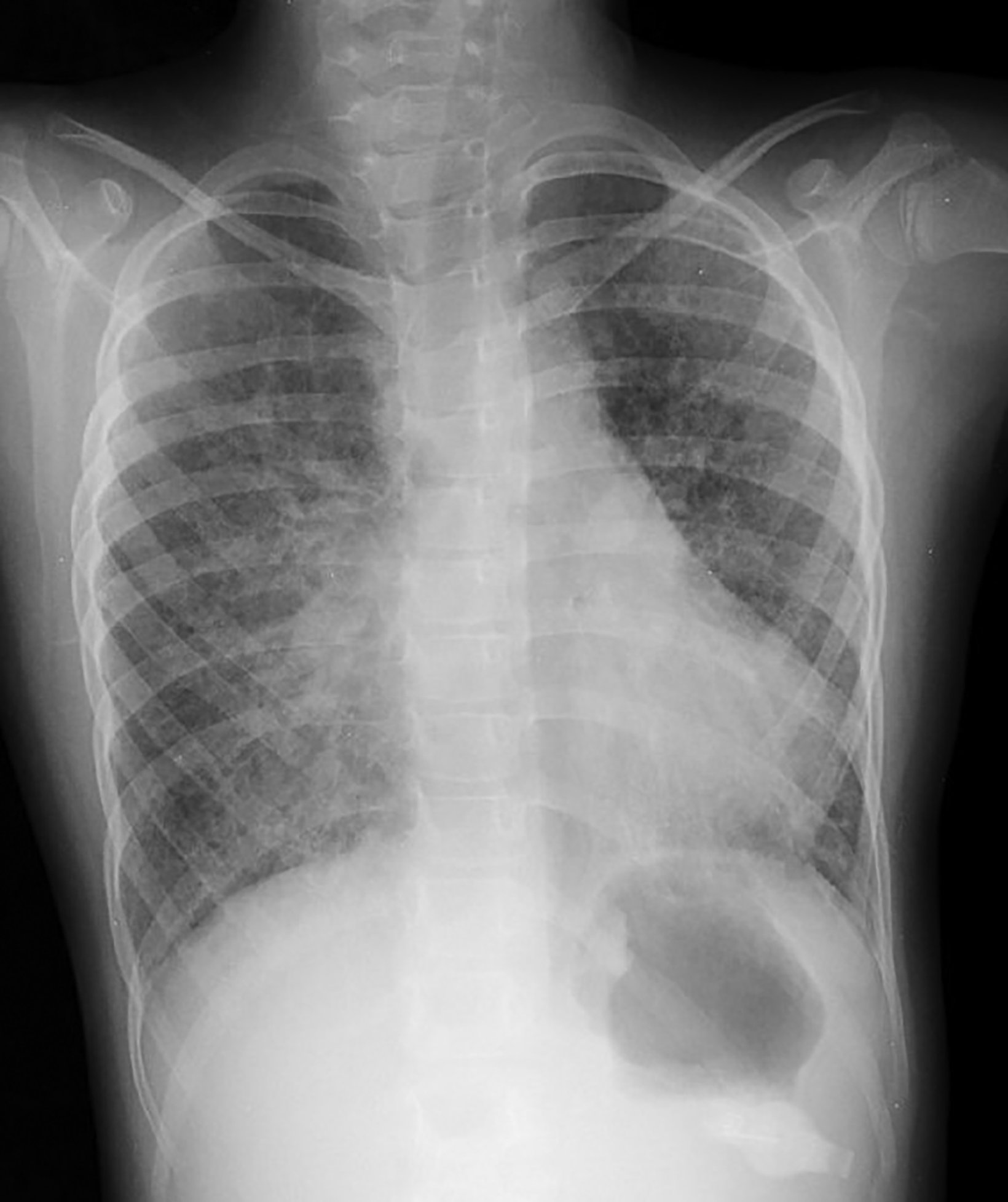
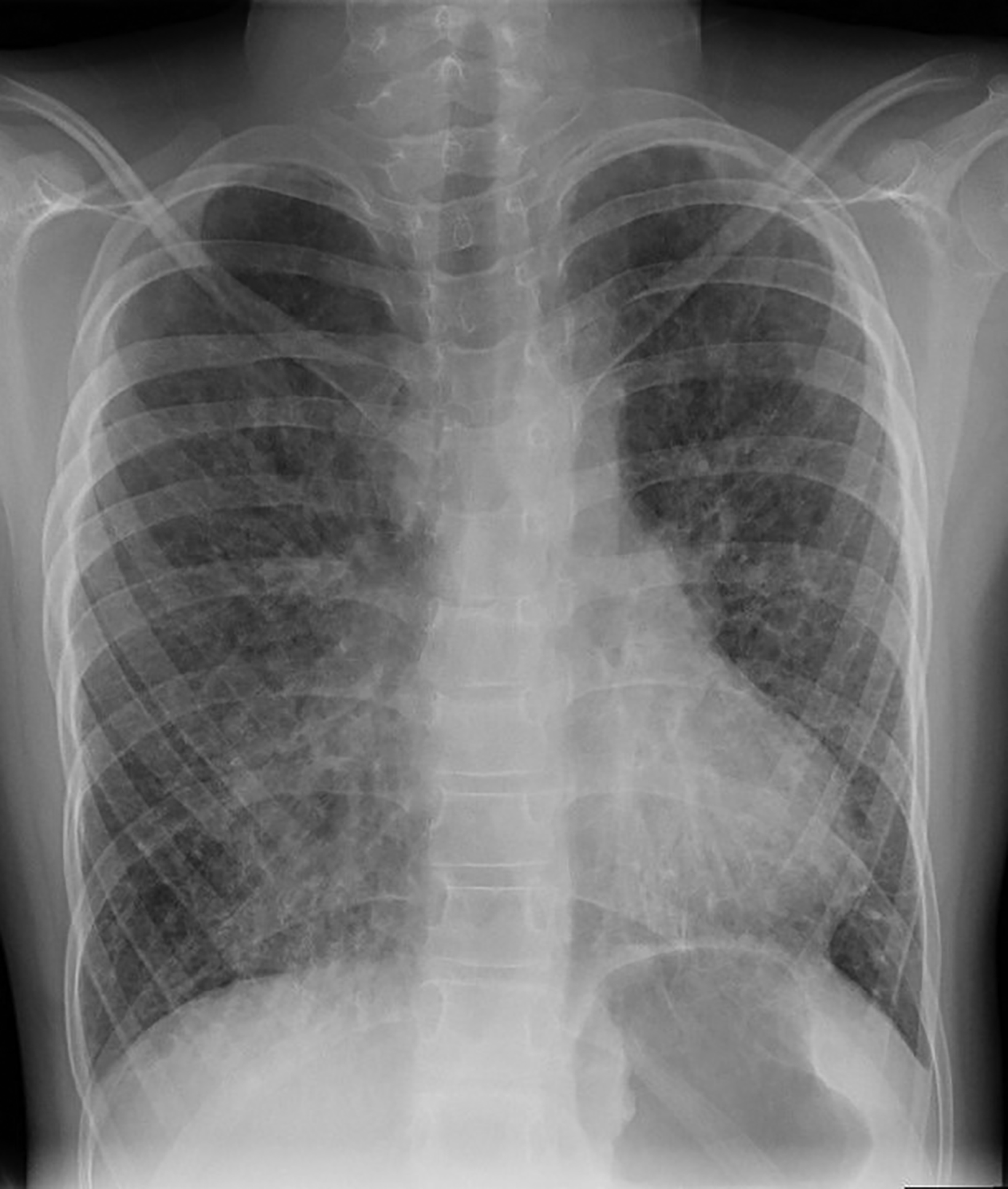
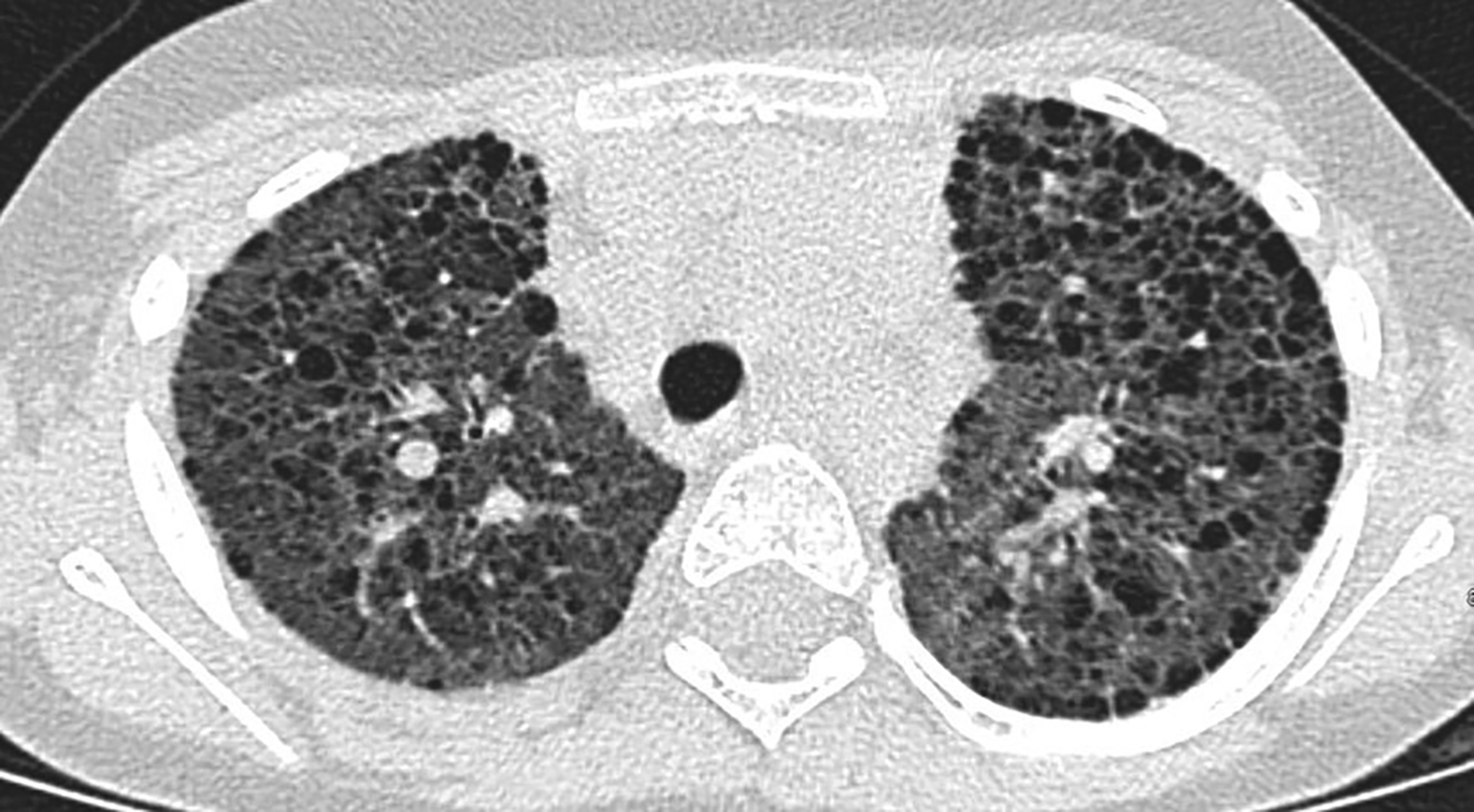
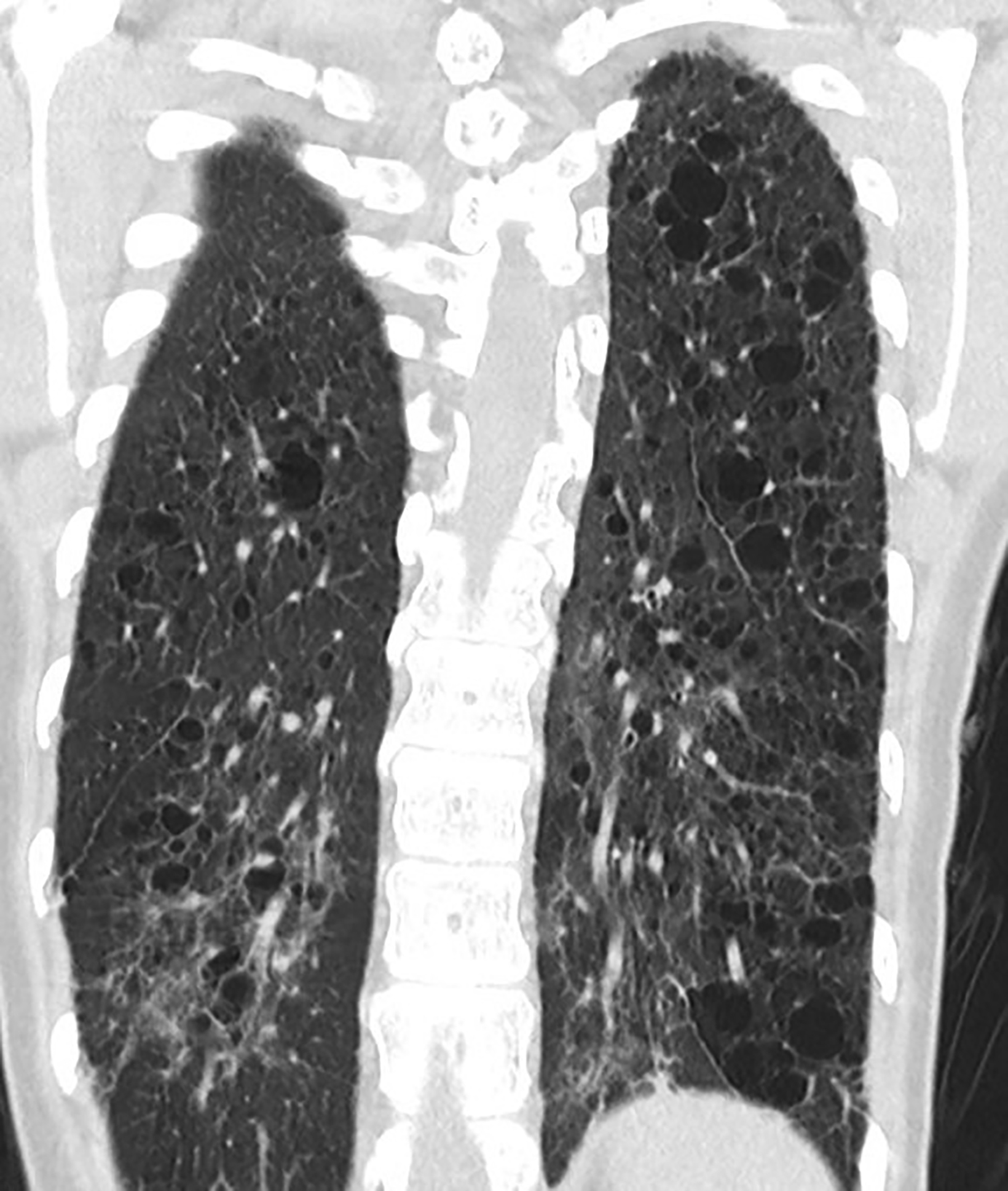
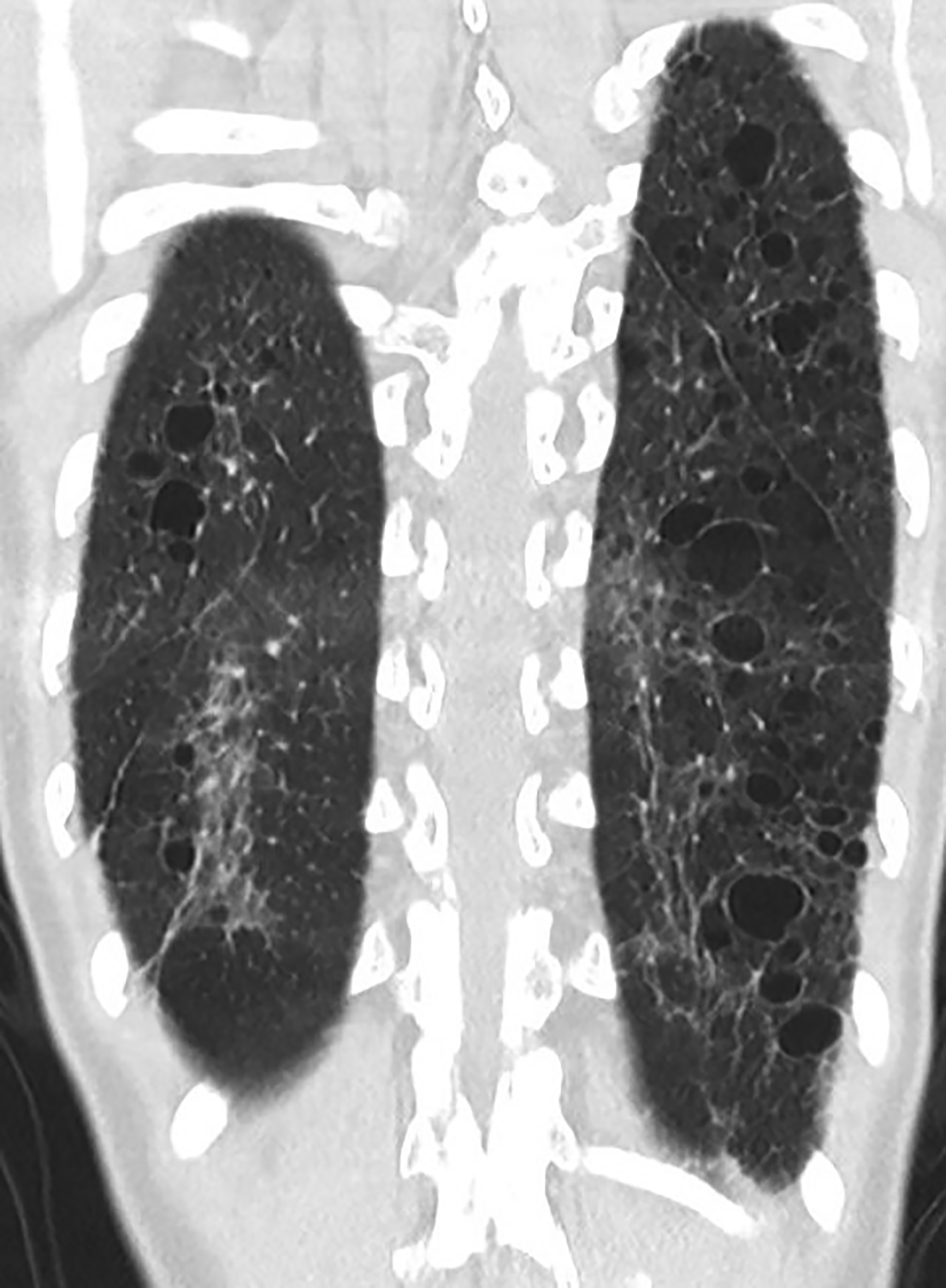
Computed tomography (CT) of the chest obtained in childhood revealed multiple, small, thin-walled cysts throughout the lungs. Follow-up CT obtained in early adolescence redemonstrated multiple, randomly distributed, thin-walled cysts in both lungs with interval new ground-glass attenuation areas in bilateral lower lobes (Figure 2). There was progressive interstitial thickening and fibrosis with age, predominantly involving bilateral lower lobes. There were no focal areas of consolidation to suggest bronchiolitis obliterans. The imaging features represent chronic cystic interstitial lung disease due to surfactant deficiency.
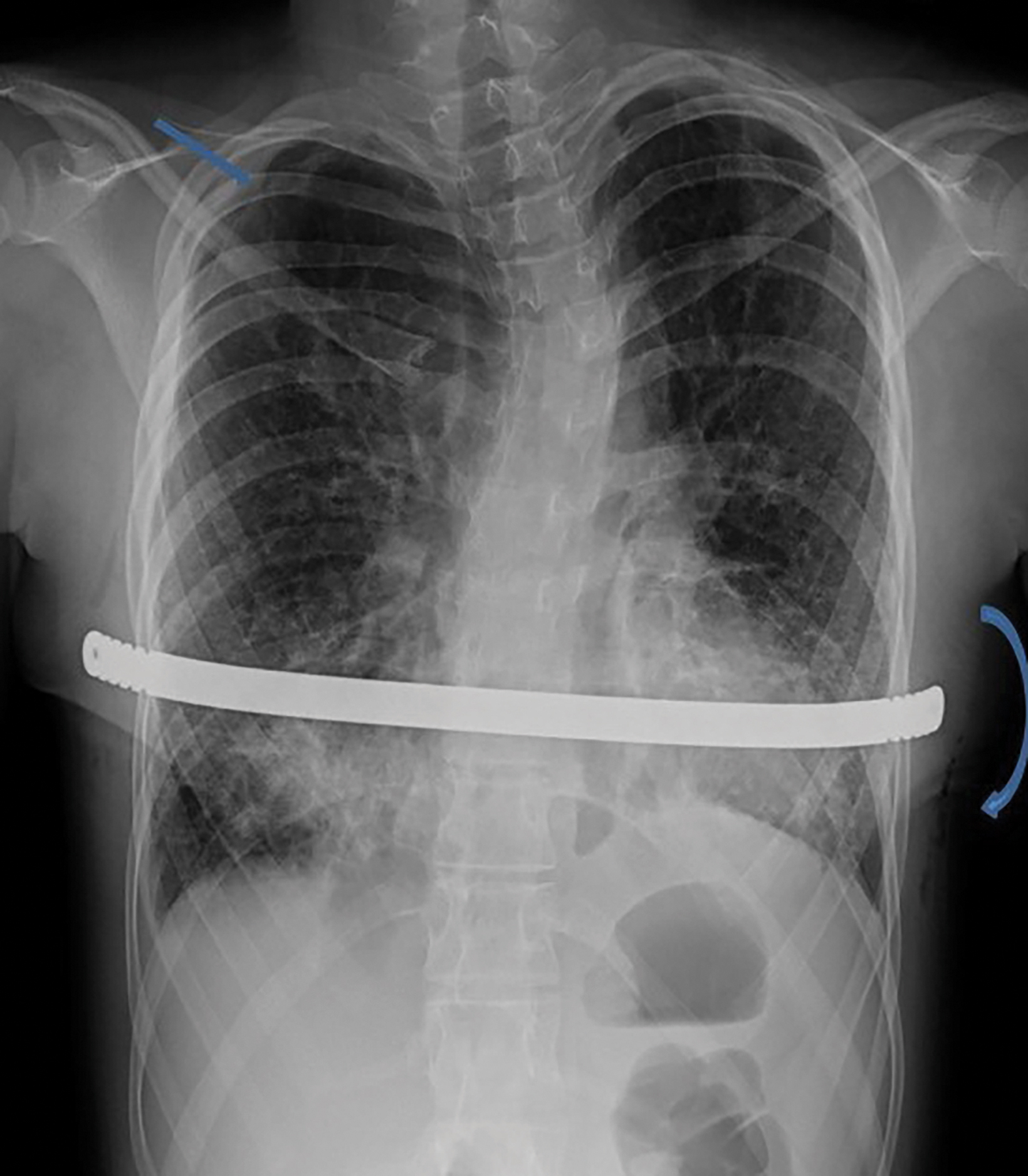
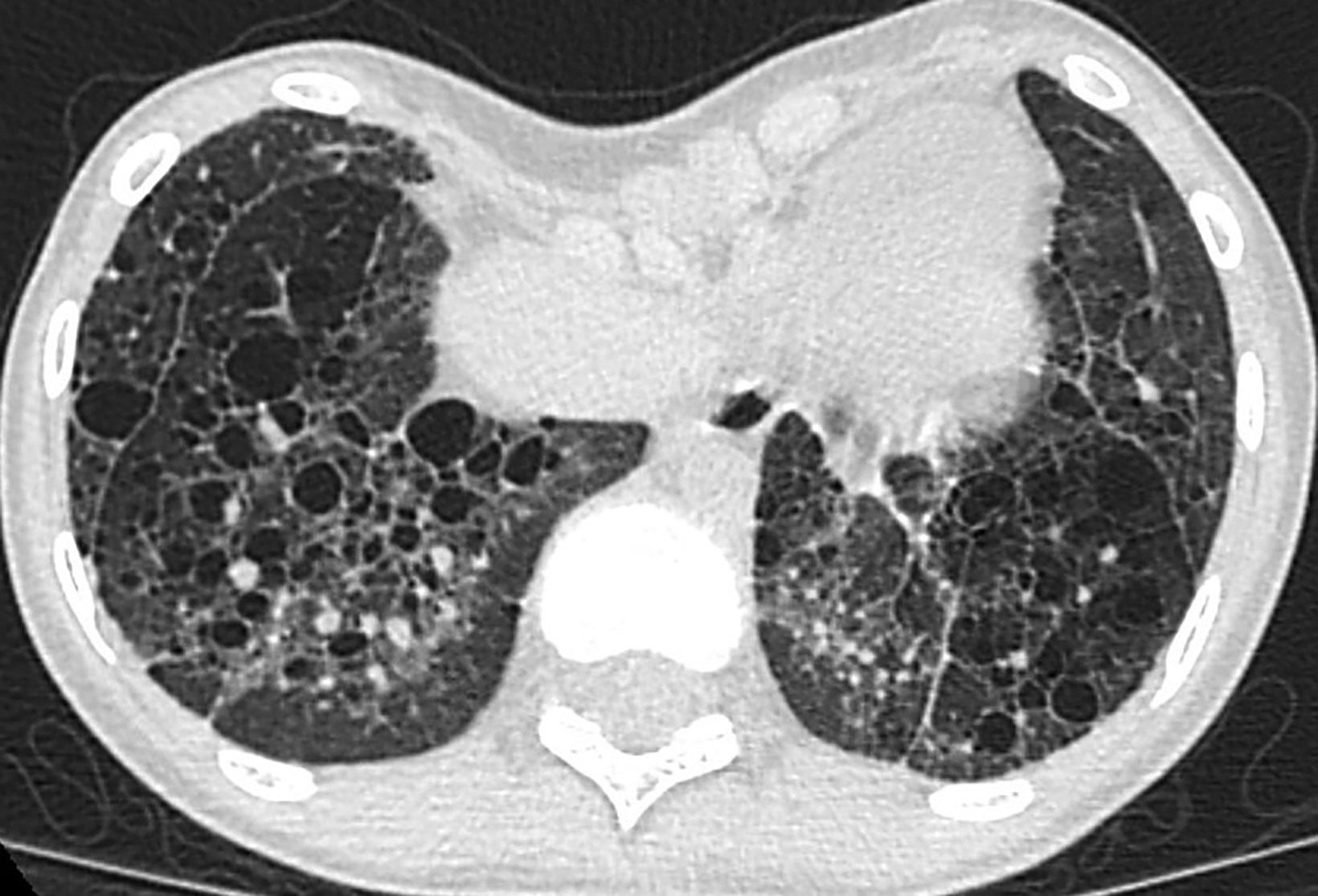
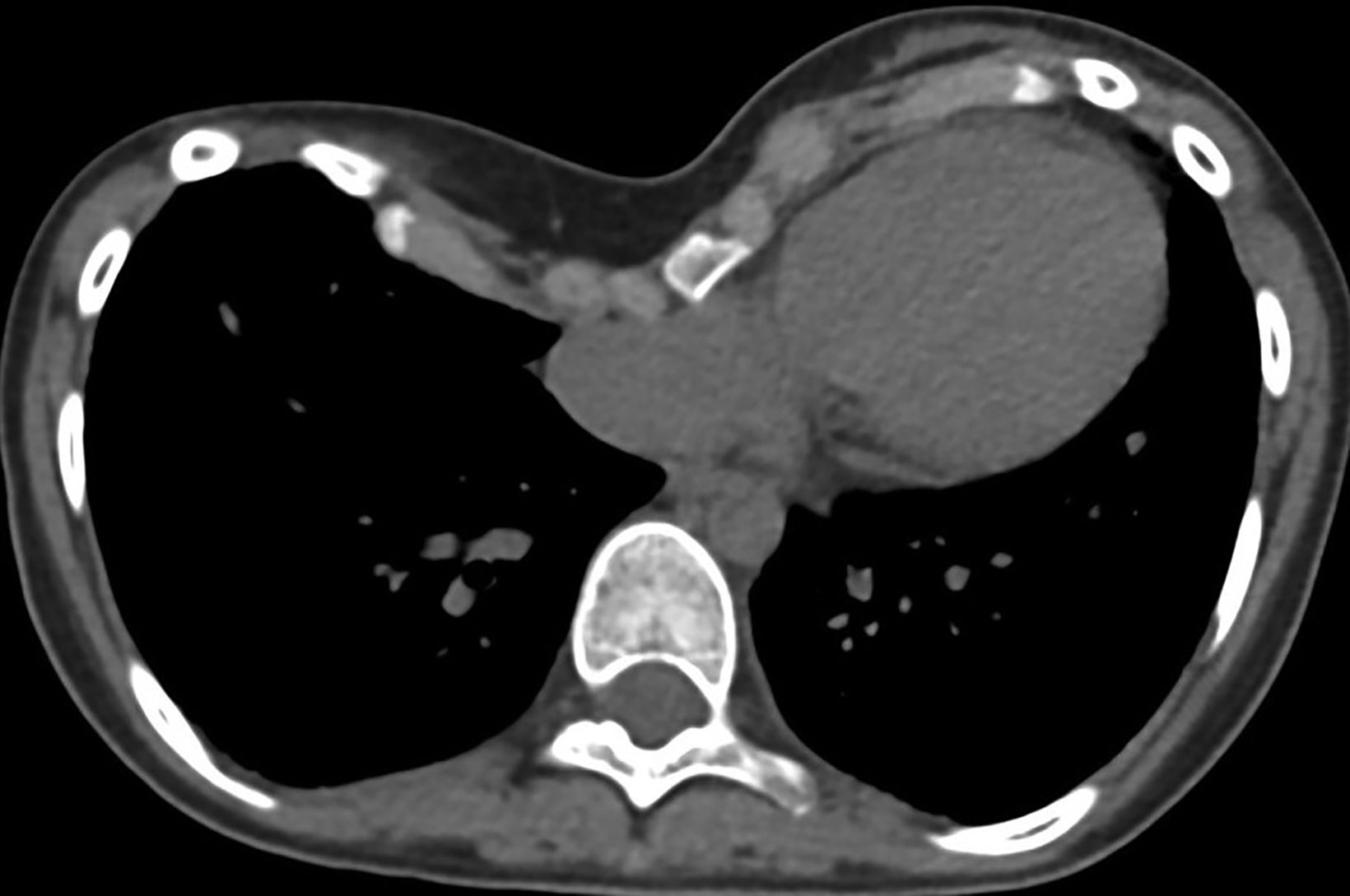
Additionally, PE deformity and thoracic spinal scoliosis developed and then worsened with age. There is a known association of PE with congenital cystic lung disease, the former being characterized by depression of the anterior chest wall, resulting in cardiopulmonary compression symptoms and restricted pulmonary ventilation disorders.1 Severity of PE is assessed by the Haller index (HI), which is calculated by dividing the maximum transverse diameter of the chest by the distance between the posterior surface of the sternum and the anterior surface of the vertebra. This is typically measured on the axial CT slice with the shortest distance between the two. An HI greater than 3.25 indicates severe PE (normal being 2.0 or less). In this patient it was 5.4 (Figure 3). Surgical correction of PE is war- ranted in symptomatic patients with a high HI. Our patient underwent a minimally invasive procedure with substernal placement of a concave bar (Nuss bar).
Owing to the patient’s tall height and PE, they were evaluated for Marfan syndrome; however, the criteria for Marfan disease were not met.
Diagnosis
Surfactant Protein C Deficiency-associated diffuse lung disease
Discussion
Childhood interstitial lung disease (ChILD), a subset of pediatric diffuse lung disease (DLD), is a heterogeneous group of rare childhood respiratory diseases. In children younger than 2 years of age, the term DLD is preferred over ChILD until other more common causes of DLD are excluded, and diagnosis of ChILD is confirmed.1 Although they are termed “interstitial lung diseases,” these conditions are not limited to the interstitium and can involve the alveoli, airways, lymphatics, blood vessels, and pleural spaces.1,2 ChILD conditions have a different etiologies to those of adults with ILD.3
The most common clinical sign in ChILD is tachypnea (75-93% of cases).2,4 Other signs include cough, failure to thrive, crackles, or wheezing. Before a child can be diagnosed with a ChILD syndrome, other causes such as congenital heart disease, congenital or acquired immunodeficiency syndromes, asthma, cystic fibrosis, primary ciliary dyskinesia, infection, and recurrent aspiration should be excluded. Three of the following four criteria should also be present: respiratory symptoms (cough, rapid breathing, difficulty in breathing, or exercise intolerance); respiratory signs (retractions, resting tachypnea, adventitious sounds, failure to thrive, digital clubbing, respiratory failure); hypoxemia; and diffuse abnormalities on chest radiograph or CT scan.2,4
ChILD associated with an autosomal dominant mutation in the SP-C gene was first described in 2001 by Nogee, et al.5 About 10% of pediatric DLD is caused by surfactant metabolic dysfunction.6,7 SP-C has a role in surfactant function to reduce surface tension and prevent end-expiratory atelectasis.2,3 Lung disease caused by different SP-C mutations varies greatly, from respiratory distress syndrome (RDS) in neonates to ILD in adults.7,8 Studies have revealed an earlier age of onset and a more prevalent failure to thrive in a group with SP-C mutations.7 Superimposed lung infection in infants, particularly respiratory syncytial virus, can result in respiratory failure and worsening interstitial lung changes.8
Imaging findings on chest radiographs include hazy granular pulmonary opacities in patchy or diffuse distribution. Lung volumes are usually normal to low; however, they may be increased with assisted ventilation. High-resolution CT findings include diffuse or patchy ground-glass opacities, consolidation, interlobular septal thickening, lung cysts, and paraseptal cysts. Regression of ground-glass opacities occurs with increasing patient age and development of lung cysts and septal thickening.1,4 Pectus excavatum may be seen in older children and is thought to be related to abnormal chest wall development in the setting of restrictive lung disease.1,5
Although the CT findings are nonspecific, CT can be helpful in several ways. It helps define the extent of disease, for following disease progression, guiding biopsy, and excluding other causes of clinical symptoms. Chest CT is also valuable for monitoring the effects of treatment, as there is a strong correlation between clinical improvement and CT findings.8
In the past, lung biopsy has been the diagnostic gold standard despite the potential complications. However, identifying the genes responsible for surfactant dysfunction disorders avoids the need for a biopsy in approximately 10% of children.7 With the broader availability of genetic testing, ILD CT imaging patterns may sufficiently suggest a diagnosis, thus avoiding the need for a surgical lung biopsy.2 Surgical lung biopsy using video-assisted thoracoscopy is recommended for infants with clinical urgency to identify the specific form of pediatric ILD or in whom other diagnostic evaluations have not yielded a specific diagnosis.2 Hence, an interdisciplinary clinical, radiological, and histopathological consensus is currently the gold standard.9,10
Conclusion
Surfactant Protein-C deficiency is a rare lung disease with highly variable age of onset, severity, and natural history. It varies greatly, from RDS in neonates to ILD in adults. An interdisciplinary clinical, radiological, and histopathological consensus is currently the diagnostic gold standard. HRCT imaging patterns of ILD may sufficiently suggest a diagnosis that can be confirmed by genetic testing and avoid the need for a surgical lung biopsy.
References
- Zucker EJ, Guillerman RP, Fishman MP, Casey AM, Lillehei CW and Lee EY. Diffuse Lung disease. In: Coley BD, Bates DG, editors. Caffey’s Pediatric Diagnostic Imaging. Elsevier Saunders: Philadelphia; 2013. pp. 595-597.
- Kurland G, Deterding RR, Hagood JS, et al. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013;188(3):376-94. doi: 10.1164/rccm.201305-0923ST. PMID: 23905526; PM-CID: PMC3778735.
- Hime NJ, Fitzgerald D, Robinson P, et al. Childhood interstitial lung disease due to surfactant protein C deficiency: frequent use and costs of hospital services for a single case in Australia. Orphanet J Rare Dis. 2014; 9:36. PMID:24642012 doi:10.1186/1750-1172-9-36
- Thacker PG, Vargas SO, Fishman MP, Casey AM, Lee EY. Current update on iInterstitial lung disease of infancy: new classification system, diagnostic evaluation, imaging algorithms, imaging findings, and prognosis. Radiol Clin North Am. 2016 Nov;54(6):1065-1076. doi: 10.1016/j.rcl.2016.05.012. Epub 2016 Aug 12. PMID: 27719976.
- Hong D, Dai D, Liu J, et al. Clinical and genetic spectrum of interstitial lung disease in Chinese children associated with surfactant protein C mutations. Ital J Pediatr. 2019;45(1):117. PMCID:PMC6714457 doi:10.1186/s13052-019-0710-2
- Mechri, M, Epaud R, Emond S, et al. Surfactant protein C gene (SFTPC) mutation-associated lung disease: High-resolution computed tomography (HRCT) findings and its relation to histological analysis. Pediatr Pulmonol.2010;45:1021-1029. https://doi.org/10.1002/ppul.21289
- McFetridge L, McMorrow A, Morrison PJ, et al. Surfactant metabolism dysfunction and childhood interstitial lung disease (chILD). Ulster Med J. 2009;78(1):7-9. PMID:19252722.
- Salerno T, Peca D, Menchini L, et al. Surfactant Protein C-associated interstitial lung disease; three different phenotypes of the same SFTPC mutation. Ital J Pediatr. 2016 Feb 29;42:23. doi: 10.1186/s13052-016-0235-x. PMID: 26925580; PMCID: PMC4772310.
- Kazzi B, Lederer D, Arteaga-Solis E, Saqi A, Chung WK. Recurrent diffuse lung disease due to surfactant protein C deficiency. Respir Med Case Rep. 2018;25:91-95. doi: 10.1016/j.rmcr.2018.07.003. PMID: 30094155; PMCID: PMC6080219.
- Szczawińska-Popłonyk A, Breborowicz A, Langfort R. Sródmiazszowa choroba płuc w przebiegu niedoboru białka C surfaktantu współistniejacego z pierwotnym niedoborem odporności - opis przypadku [Interstitial lung disease in the course of the surfactant protein C deficiency coexisting with the primary humoral immunodeficiency - case study]. Pneumonol Alergol Pol. 2010;78(3):244-7. Polish. PMID: 20461694.
Citation
A C, PK S,. Surfactant Protein C Deficiency-associated Diffuse Lung Disease. Appl Radiol. 2023;(1):42-45.
February 1, 2023