Orbital Rhabdoid Tumor
Case Summary
An infant presented with a 2-week history of a bulging left eye, with proptosis and entrapment. Orbital CT and MRI revealed a left orbital mass. Biopsy revealed that the mass was a rhabdoid tumor (RT), and the patient subsequently underwent chemotherapy. Treatment significantly decreased the mass.
Imaging Findings
Orbital CT with contrast ( Figure 1 ) and orbital MRI without and with contrast ( Figure 2 ) revealed a heterogeneously enhancing 3.6 × 1.7-cm retrobulbar mass in the left orbit. No intracranial extension was noted. CT of the chest, abdomen, and pelvis showed no metastatic disease.
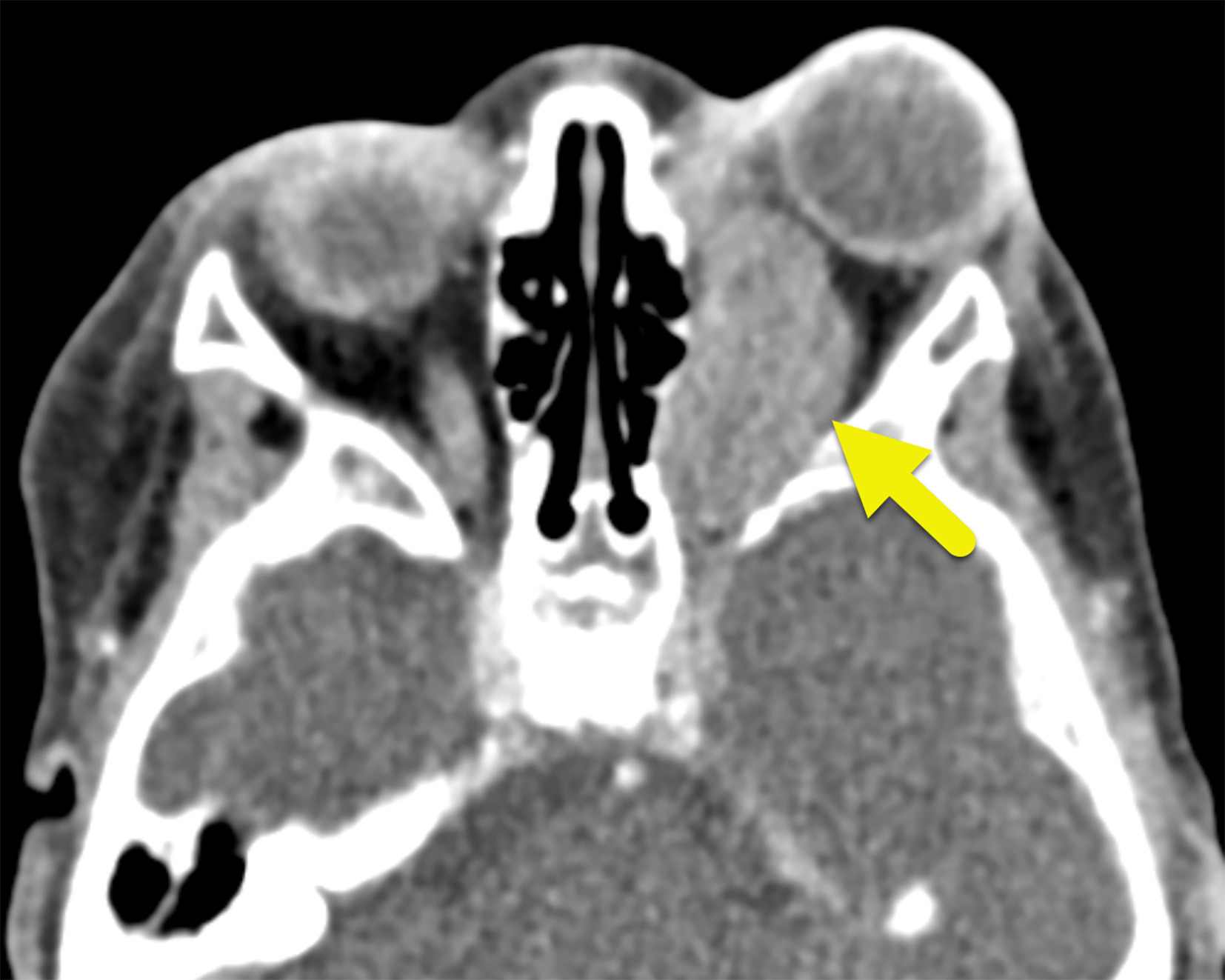
Axial contrast-enhanced CT shows a mildly enhancing mass (arrow) of the left orbit causing proptosis.
Axial T1-weighted MRI (A) shows an isointense orbital mass (arrow) causing proptosis. Axial T2-weighted MRI (B) shows the lobulated mass (arrow) to be heterogeneous and mostly hypointense with small hyperintense foci. T1-weighted postcontrast MRI (C) showing the mass (arrow) to have mild peripheral enhancement.

Diagnosis
Rhabdoid tumor.
The differential diagnosis includes other similar round-cell tumors such as neuroblastoma and rhabdomyosarcoma.
Discussion
RT of the orbit was initially described as a sarcomatous variant of Wilms tumor in 1978. However, since then it has been recognized as a distinct entity.1 RTs have a wide phenotypic range and can be present in many parts of the body. According to a population study using data from the Surveillance, Epidemiology, and End Results (SEER) program, RTs most commonly arise in the central nervous system (CNS) (35%) and kidneys (20%), with the remaining 45% located extracranially.2, 3 An RT can occur anywhere in the brain or spine. CNS RT, also called atypical teratoid rhabdoid tumor, most commonly arises within the posterior fossa.4 - 6
Histologically, RTs have polygonal cells with eccentric nuclei and hyperchromatic nucleoli with vimentin and epithelial membrane antigen positivity.2, 7, 8 Additionally, RTs are united by a loss-of-function mutation in the SMARCB1 or, less commonly, SMARCA4 tumor suppressor gene.8 The presence of a mutation is diagnostic of MRT in the context of poorly differentiated malignant rhabdoid cells.8
Orbital RTs are rare and typically occur in infants, with a mean age of diagnosis of 10.37 months ± 14.5 months. The tumors account for less than 1% of all RTs and occur in less than 1 in a million people. Diagnosis is most often made in children 11 to 18 months old.3, 7 Patients present with rapidly progressing proptosis, preauricular/submandibular lymphadenopathy, eyelid swelling, and loss of light reaction in the eye in more advanced cases.7 - 9 Orbital RTs are not always confined to the orbit. CNS involvement has been linked with worse outcomes and poses a problem for the possibility of resection.10 Unfortunately, orbital RTs have a poor prognosis, with a 3-year survival rate of 9%.6
MRI is the most useful modality to assess the disease extent owing to its ability to assess intracranial extension and optic nerve involvement. CT is helpful when an MRI cannot be obtained or when bone destruction is suspected. Orbital RT appears as a heterogeneous retrobulbar, intraconal mass causing proptosis on MRI. It is hypo- to isointense on T1-weighted images, iso- to hyperintense on T2-weighted images with areas of edema, calcification, and hemorrhage, and inhomogeneously enhances after the administration of gadolinium-based contrast media. If CT is performed, the mass appears as a heterogeneous, hypodense solid mass. The tumor does not typically enhance after the administration of iodinated contrast media. CT of the chest and CT or MRI of the abdomen and pelvis are frequently performed to assess the presence of metastatic disease.
Treatment guidelines are not well established for this rare disease, but multimodal approaches have been used with some success in decreasing tumor burden. These approaches include combination chemotherapy, radiotherapy, and/or stereotactic radiosurgery.9 Most tumors are ultimately resected and the globe enucleated.
Conclusion
Orbital RT is a rare tumor that presents with rapid progression of proptosis and eye swelling. The tumor is typically characterized via imaging with CT and MRI and diagnosed via biopsy. Patients are treated with a multimodal approach. Orbital RTs have a poor prognosis, and continued improvements in therapy are needed.
References
Citation
Valbuena JV, Towbin RB, Morgan D, Schaefer CM, Towbin AJ. Orbital Rhabdoid Tumor. Appl Radiol. 2024;(5):14 - 16.
doi:10.37549/AR-D-24-0017
October 1, 2024