Neurosarcoidosis
Images
Case Summary
A middle-aged patient presented with an 8-week history of progressive right-sided visual decline and eye pain. She also reported left truncal paresthesias, imbalance, bilateral tinnitus, and dizziness. The patient’s family history was notable for a vestibular schwannoma in their parent. On exam, the patient had decreased sensation to pinprick over the left T6 to T10 levels and mildly diminished proprioception in bilateral lower extremities. Visual testing showed a right inferior arcuate defect with papilledema and 2 mm proptosis. Visual acuity was asymmetrical. A right afferent pupillary defect was noted, and optic nerve function was asymmetric.
Imaging Findings
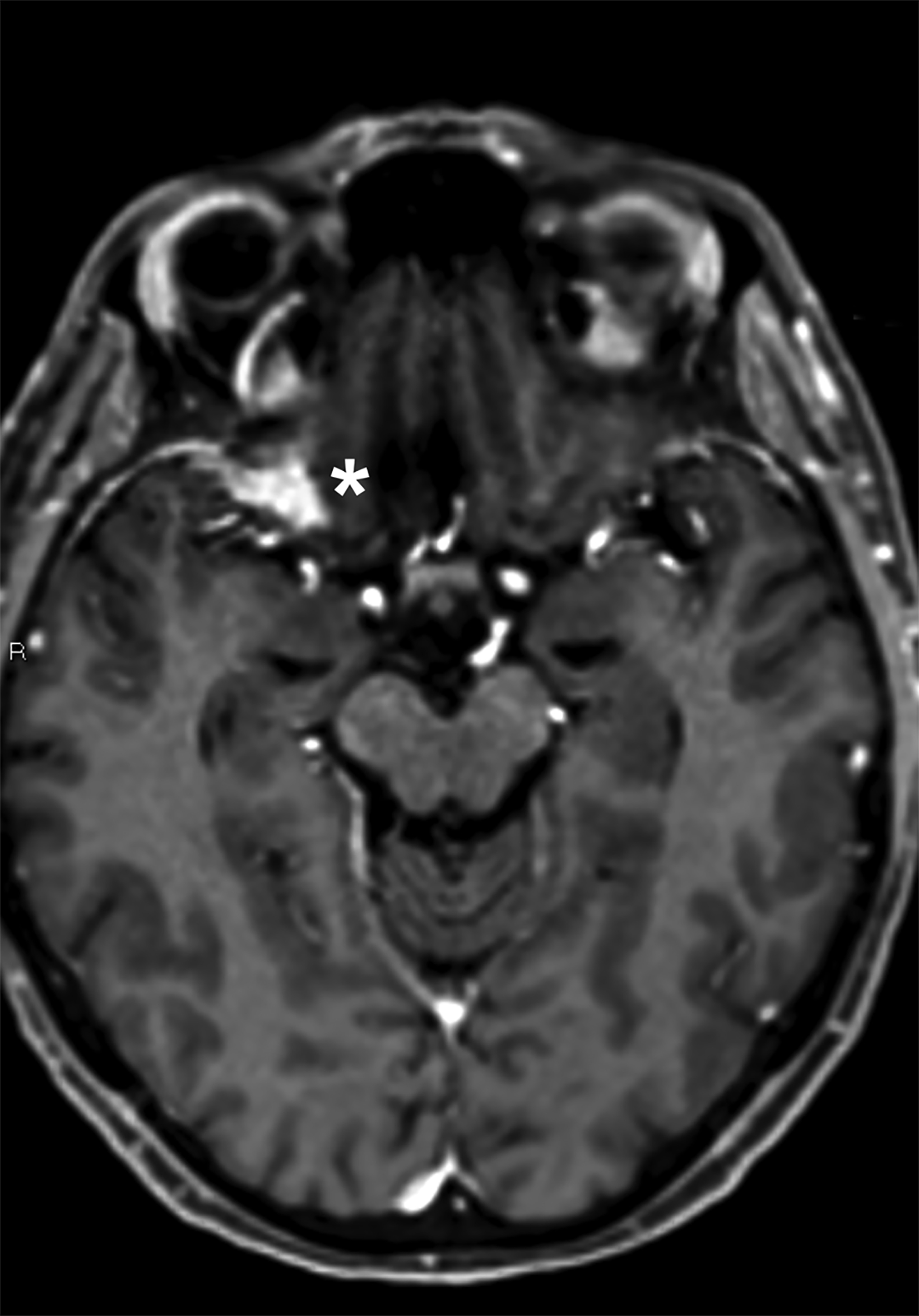
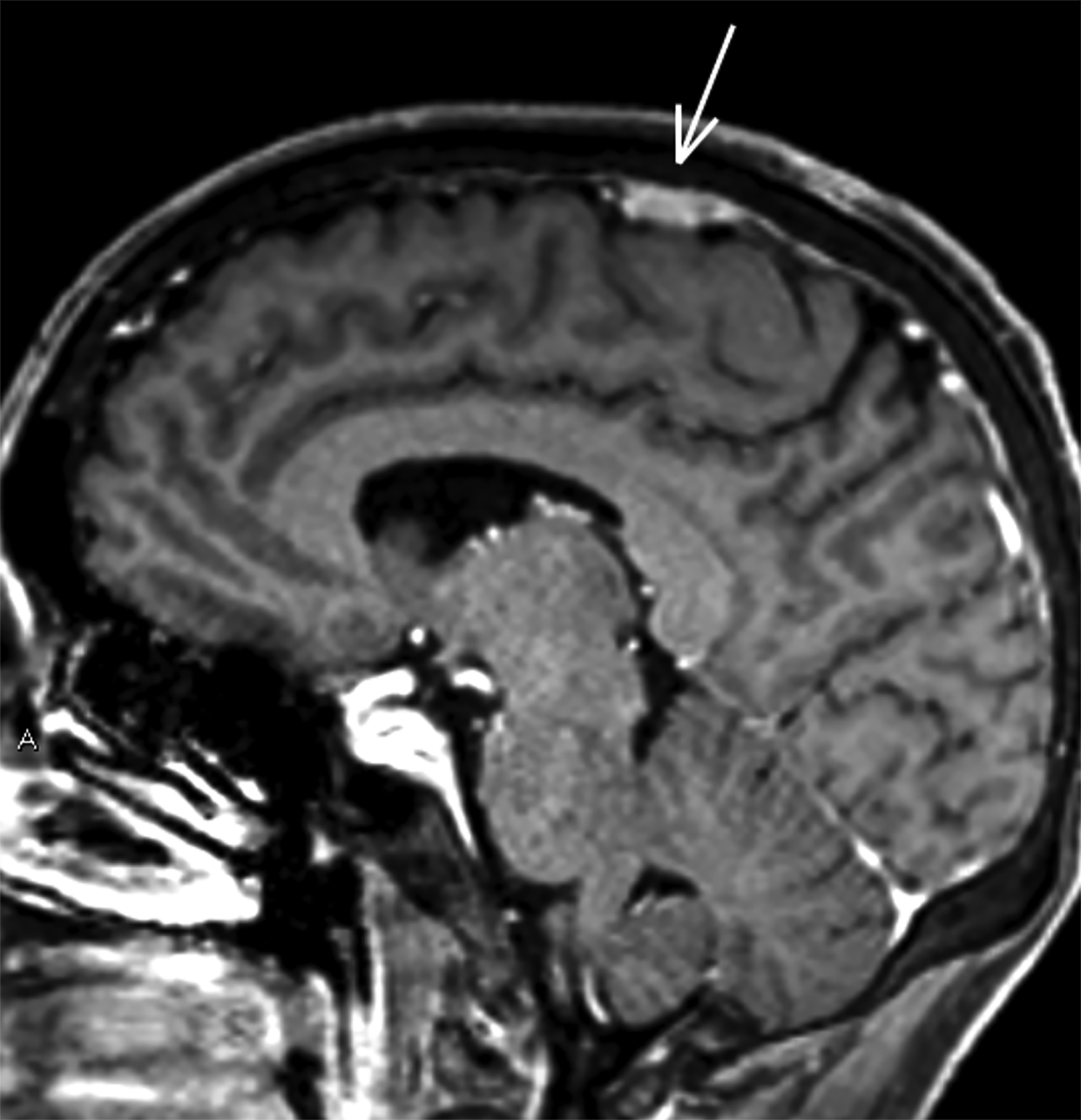
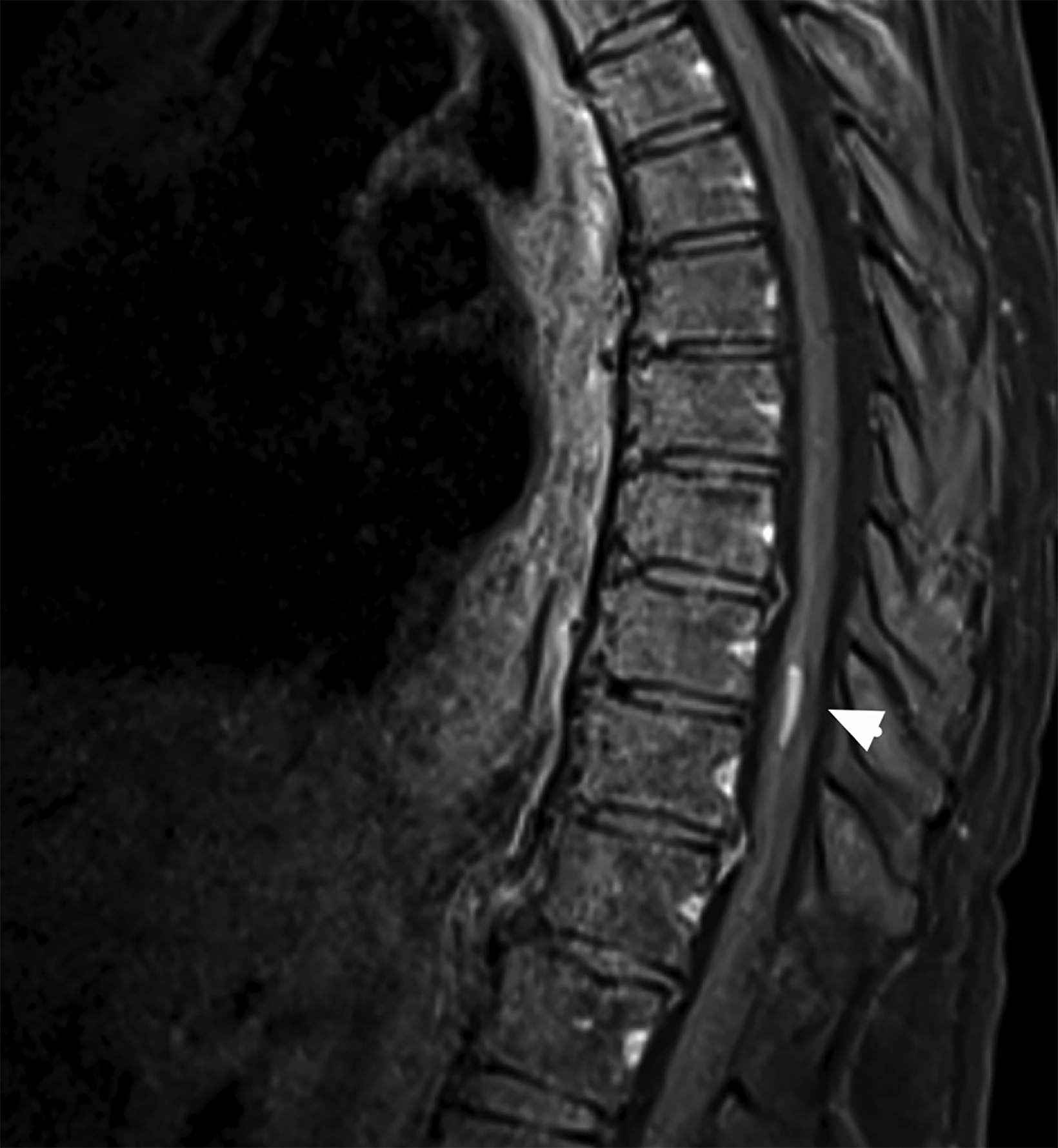
Magnetic resonance imaging (MRI) revealed homogeneously contrast-enhancing lesions along the lesser wing of the right sphenoid and left parasagittal vertex with dural “tails.” MRI of the thoracic spine showed a contrast-enhancing lesion centrally within the spinal cord at the T9-10 level with mild perilesional edema (Figure 1). Given the multiple extra-axial dura-based lesions (possible meningiomas), enhancing lesion within the thoracic spinal cord (possible ependymoma), and family history of vestibular schwannoma, the differential diagnosis included neurofibromatosis type 2 (NF2). Computed tomography of the chest, abdomen, and pelvis revealed only a few slightly enlarged hilar region lymph nodes.
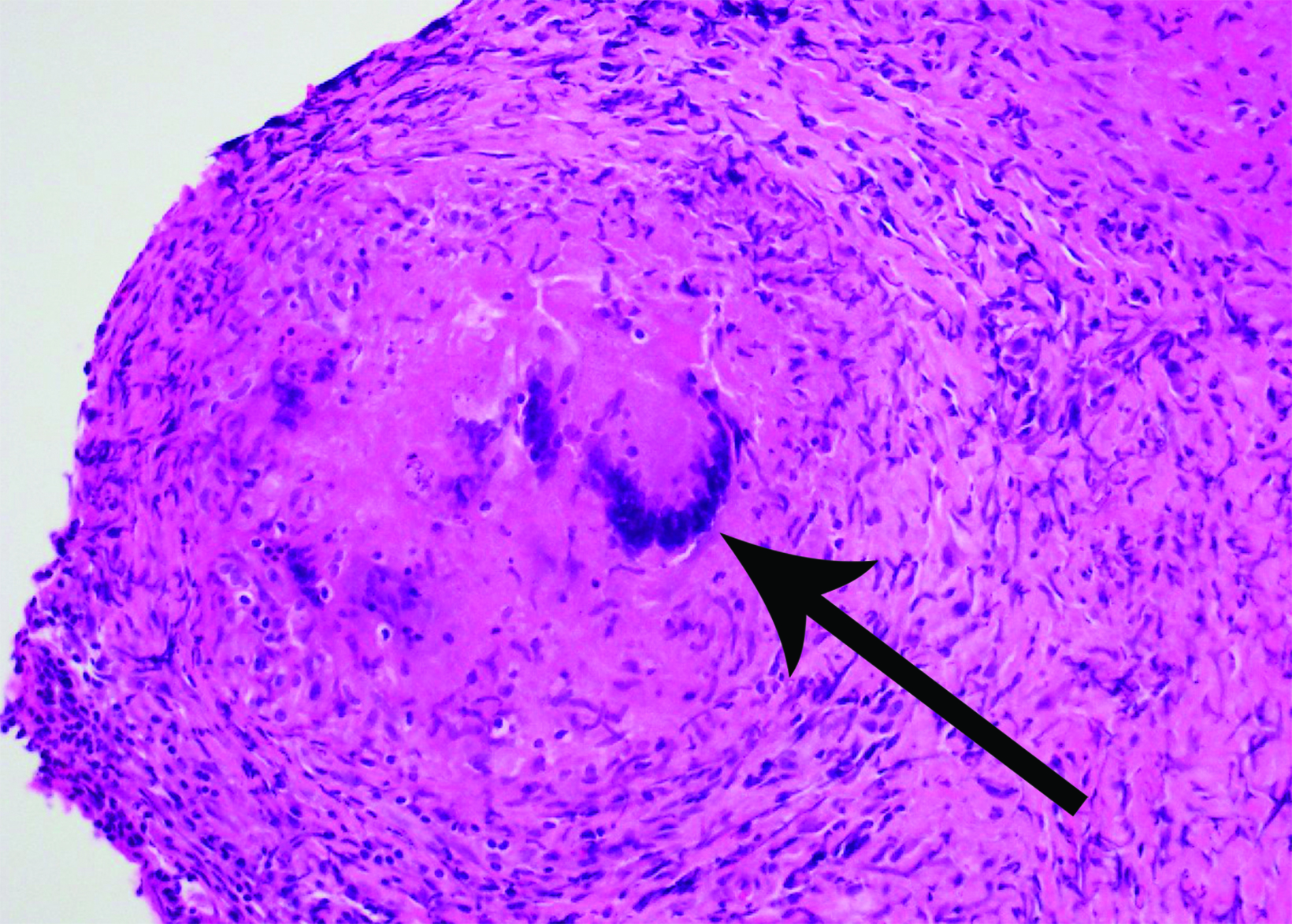
A lumbar puncture showed normal opening pressure, normal cerebrospinal fluid profile, normal IgG index and negative oligoclonal bands. Serum angiotensin-converting enzyme was normal. A mediastinal lymph node biopsy was nondiagnostic, showing rare noncaseating granulomas but was insufficient for diagnosis. Biopsy was recommended via a right pterional craniotomy. At surgery, the lesion was noted to be pale and firm; histopathology demonstrated noncaseating granulomatous inflammation (Figure 2).
Diagnosis
Neurosarcoidosis. Differential diagnosis includes neurofibromatosis type 2, given the extra-axial right sphenoid wing and left parasagittal vertex lesions mimicking meningiomas, as well as the intramedullary thoracic lesion mimicking ependymoma, especially in a patient with a family history of schwannoma.
Discussion
Sarcoidosis is a granulomatous disease that affects 11 in 100,000 Caucasians and 36 in 100,000 African Americans in the United States.1 The respiratory and lymphatic systems are most commonly involved; however, central nervous system (CNS) involvement is seen in roughly 5-13% of cases.2 The average age at diagnosis of neurosarcoidosis is 33-41 years, although individuals of all ages may be affected.2 Women and African Americans are affected more commonly, similar to systemic sarcoidosis.1
Signs and symptoms of neurosarcoidosis can mimic a variety of disorders, as it can affect the hypothalamus, leptomeninges, cranial nerves, spinal cord, peripheral nerves, and muscle tissue. Cranial nerves are most commonly involved, especially VII and II, leading to facial weakness, decreased visual acuity, retrobulbar pain and visual field deficits.1,3 Other cranial nerve involvement can lead to auditory or vestibular dysfunction (VIII), anosmia (I) or ophthalmoplegia (III, IV, VI).3,4
Intracranial neurosarcoidosis may also cause headaches, ataxia, nausea and vomiting, seizures, and cognitive dysfunction.5 On cross-sectional imaging, it is known as one of “the great mimickers.” Leptomeningeal involvement can be seen in up to 40% of cases and is usually associated with elevated cerebrospinal fluid protein and mononuclear inflammatory cells.6 In the spinal cord, 15-28% of patients have MRI-visible lesions which may be extra- or intradural, extra- or intramedullary, and/or arachnoidal adhesions.5
Diagnostic criteria for neurosarcoidosis include histologic confirmation of noncaseating granulomas, compatible clinical scenario, and confirmatory imaging studies with exclusion of other diagnoses, including infectious agents.
Neurosarcoidosis can also mimic common benign skull base neoplasms. Carlson et al described 305 patients with presumed benign skull-base tumors, which ultimately were found to be sequelae of neurosarcoidosis.9 In their series, meningeal thickening that mimicked meningiomas or schwannomas occurred frequently in the cavernous sinus, sphenoid ridge, and temporal bone. Bilateral presumed vestibular schwannomas were observed in 6 cases in their series.
Neurofibromatosis type 2 is an autosomal-dominant tumor predisposition syndrome that occurs in approximately 1 in 25,000 individuals.7 While approximately half of cases are familial, de novo mutations represent the other half, obviating the need for a family history of NF2 for diagnosis. NF2 results from a germline mutation in the NF2 tumor suppressor gene on chromosome 22q12, causing schwannomas, meningiomas, and spinal cord ependymomas. The most widely utilized clinical diagnostic schema for NF2 diagnosis, the Manchester NF2 clinical criteria, calls for at least 2 additional NF2 findings (nonvestibular cranial or peripheral nerve schwannoma, spinal cord ependymoma, retinal hamartoma/epiretinal membrane, or cataracts) in patients whose principal findings are multiple meningiomas.8
Our case underscores the need to be familiar not only with the typical imaging findings in patients with NF2, but also with the workup of conditions that may mimic NF2, such as neurosarcoidosis. Accurate and clinically available clinical biomarkers remain necessary for ready diagnosis of this entity without histopathologic confirmation of surgically acquired specimens, especially when surgery carries elevated risks.
Conclusion
Neurosarcoidosis is a granulomatous disease that may masquerade as a number of clinical disorders, owing to its varied involvement of cranial nerves, leptomeninges, peripheral nerves, and the spine. A high index of suspicion should be maintained in patients presenting with compatible clinical scenarios and imaging findings. Pathological confirmation of noncaseating granulomas and absence of infectious causes are necessary for definitive diagnosis and treatment of this clinical entity.
References
Citation
DT A, M S, T W, MB L, A A,. Neurosarcoidosis. Appl Radiol. 2023;(2):42-44.
March 7, 2023