Langerhans cell histiocytosis
Images
![]()
CASE SUMMARY
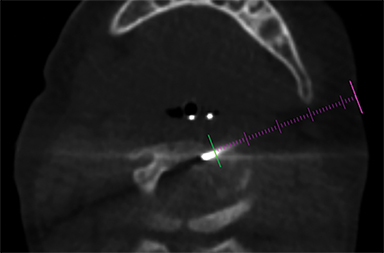
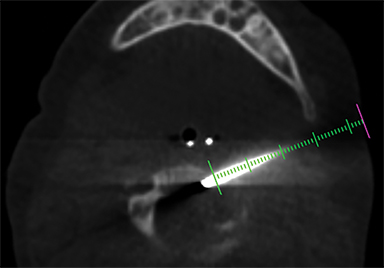
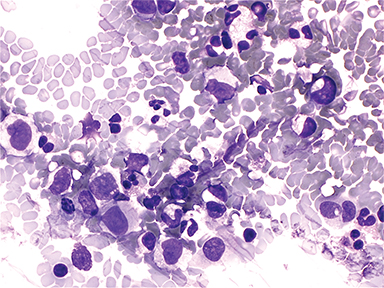
A 15-month-old boy presented to our institution with a history of persistent neck pain. A diagnosis of bilateral mastoiditis was made a few weeks earlier after the child had refused to move his neck. He was started on a 10-day course of antibiotics, but returned after completing treatment owing to worsening symptoms. Cervical spine radiographs and CT revealed a lytic lesion of the C2 vertebral body (Figure 1). Prior to percutaneous biopsy, MRI (Figure 2) and PET CT (Figure 3) scans were obtained to confirm the region of maximal activity for diagnosis and biopsy route planning. Cone beam-guided, fine-needle aspiration (FNA, Figure 4) provided cells for cytology, and immunostains performed on cell block material prepared from the needle rinsings (Figure 5) confirmed the diagnosis.
IMAGING FINDINGS
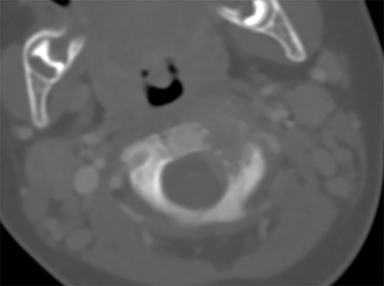
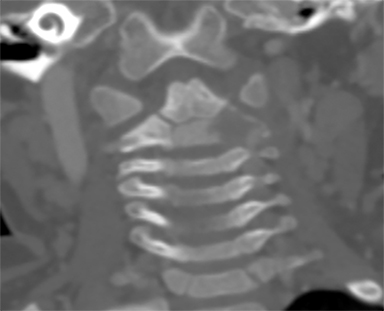
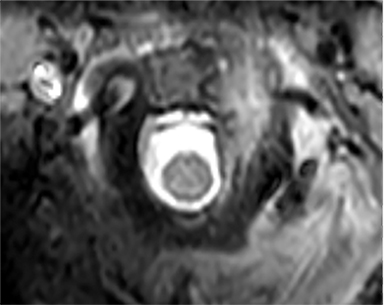
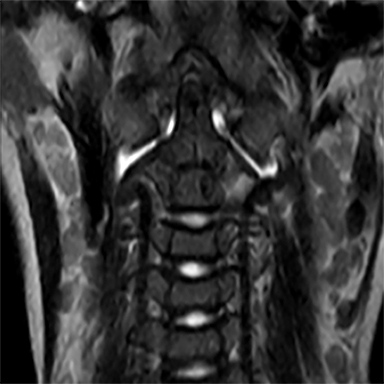
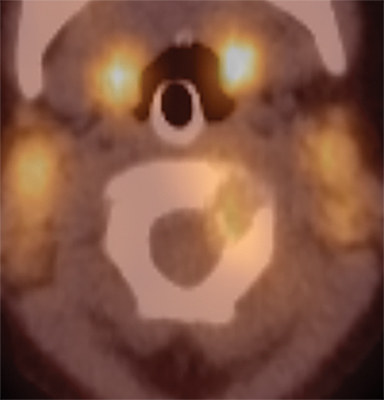
Contrast CT (Figure 1) revealed a lytic lesion with enhancing soft tissue involving the epidural space at the left lateral aspect of the C2 vertebral body. Subsequent MRI (Figure 2) showed an enhancing mass in the lateral C2 vertebral body, with slight narrowing of the spinal canal and lateral displacement of the left vertebral artery. PET CT (Figure 3) confirmed that there was increased FDG uptake in the area of the lytic lesion.
DIAGNOSIS
Langerhans cell histiocytosis (LCH).
The differential diagnosis includes leukemia/lymphoma, osteosarcoma, Ewing sarcoma, bony metastasis and osteomyelitis.
DISCUSSION
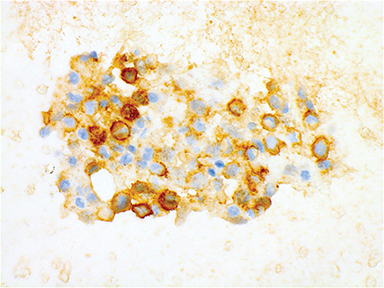
Langherhans cell histiocytosis is a rare disorder characterized by accumulation of a type of dendritic antigen-presenting cell, the Langerhans cell, resulting in protean clinical manifestations ranging from solitary bone involvement to multivisceral infiltration.1 The true nature of LCH as either neoplastic or reactive has historically been steeped in controversy, with recent advances confirming the former in many cases (some cases of isolated pulmonary LCH in adults whose mainstay of treatment is smoking cessation may be an exception). The most common mutation seen in LCH is in the BRAF gene, with less common mutations occurring in TP53, RAS, and MET genes. About one-half of LCH cases harbor BRAFV600E point mutations, which can be helpful diagnostically when looking for systemic involvement (looking for circulating cell free DNA with this mutation).2 The annual incidence of LCH in children younger than 15 years of age is approximately 8.9/1,000,000, with a median age at diagnosis of 3 years.3 LCH can affect nearly any organ, but most commonly affects bone (80% of cases) in pediatric populations.3 Immunohistochemical staining with CD1a or langerin (CD207; which is more specific than but not as widely available as CD1a) is necessary to confirm the diagnosis, with S-100 positivity lending further support (Figure 5).3
Normal Langerhans cells are found in the epidermis, representing 1-2% of all epidermal cells. It is in the epidermis where the Langerhans cell performs its role of immunosurveillance, extending dendritic processes into mucosal sites to maximize the surface area over which they can sample antigens. It is at this point when the Langerhans cell migrates to the lymph node and presents these antigens to the T cell, producing cytokines and chemokines and upregulating cytokine receptors and costimulatory molecules in the process. It is thought that aberrant expression of chemokine receptors contributes to this pathologic accumulation of Langerhans cells, allowing for inappropriate migration into various tissue types.4
Unifocal, unisystem LCH is a benign subtype of LCH hallmarked by infiltration of pathologic Langerhans cells, often mixed with eosinophils and lymphocytes confined to a specific anatomic site. Eosinophils are often times the predominant cell type in the infiltrate. It is most frequently encountered within the medullary cavity of bones such as the skull, ribs, and femur, but lesions can also less commonly arise in the skin, lungs, or stomach. Clinical symptoms of this subtype of LCH arising in the bone include pain, tenderness, or pathological fractures. A common manifestation in young children presents with multiple erosive bony masses that can expand into surrounding soft tissue.5 Unifocal LCH occurs relatively infrequently in the spine; moreover, the incidence in specific spinal segments occur most frequently in thoracic regions, followed by lumbar, then cervical.5 Among the pediatric cervical cases, previous studies suggest it is most common in C3-C5 segments, with the body of the vertebra being the most commonly affected site. The most often presenting symptoms are usually pain and restricted range of motion or torticollis. Neurological symptoms are very rarely seen with spinal involvement, but do occur.5
Plain film radiographs of the affected areas in bony LCH will typically reveal lytic lesions, with well-defined borders in more indolent lesions and poorly-defined borders in more aggressive forms. On CT, lytic lesions with cortical disruption are often evident, and soft-tissue involvement may occur. On magnetic resonance, most bone lesions are hypo-intense on T1-weighted images, hyper-intense on T2-weighted images, and show more soft tissue involvement.6 Vertebral body involvement often results in a near collapse of the affected segment, resulting in a characteristic ‘vertebra plana’ appearance seen on lateral view radiography.6
The differential diagnosis for lytic bone lesion is broad. Other disease processes that can cause such a lesion in the pediatric population include malignancy (osteosarcoma, Ewing sarcoma, leukemia, lymphoma) and infection. A distinctive feature that favors unifocal, unisystem LCH involvement of the spine over infection is preservation of the disc space on MR.6 Definitive diagnosis is achieved with demonstration of CD1a, CD207, and S-100 positivity on IHC in addition to the aforementioned radiographic features.
The prognosis of LCH varies depending on the extent of disease and organ involvement. For unifocal unisystem involvement, LCH follows a fairly benign course, has an excellent prognosis, and tends to resolve with minimal treatment. Localized granulomas often respond well to simple surgical excision. Often times bony lesions resolve spontaneously. Patients with more diffuse system involvement usually require systemic chemotherapy. Common agents include, etoposide, methotrexate, vinca alkaloids, and prednisone.7 Recently, targeted therapy using BRAF inhibitors such as vemurafenib have shown to be efficacious and should be considered in refractory cases of LCH harboring the BRAF mutation.8
CONCLUSION
Langerhans cell histiocytosis is a disorder of dendritic cells. It can cause varying clinical manifestations and affects patients of all ages. It most commonly affects the skeletal system in children. Pathologic diagnosis of LCH is established by identifying lesional Langerhans cells in an appropriate cellular milieu. Unisystem, unifocal LCH is a subtype of LCH with isolated system involvement. Radiological findings include bony lytic lesions involving the vertebral body and ‘vertebra plana’ appearance seen on lateral view. This particular subtype of LCH tends to follow a more indolent course, and has an excellent prognosis. Treatment options include surgical resection, systemic chemotherapy, and BRAF inhibitors. This particular case of LCH occurring in the C2 vertebral body allowed us to implement a novel interventional approach by sampling the lesion trans-orally; this proved to be both challenging and rewarding and paves the way for future interventions in similar cases. Collaborative efforts with a cytopathologist ensured a successful procedure and a safe outcome for the patient.
REFERENCES
- Emile, J, Abla, O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672-2681.
- Héritier, S, Hélias-Rodzewicz, Z, Lapillonne, H, et al. Circulating cell-free BRAFV600E as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol. 2017; 178:457-467.
- Monsereenusorn, C, Rodriguez-Galindo C. Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis. Hematology/oncology clinics of North America. 2015; 29(5):853-873.
- Kumar, V, Abbas, AK, Aster, JC. Langerhans Cell Histiocytosis. Robbins and Cotran Pathologic Basis of Disease, 9th ed. 2015:621-622.
- Bertram, C, Madert, J, Eggers, C. Eosinophilic Granuloma of the Cervical Spine. Spine. 2002;27(13):1408-1413.
- Azouz, EM, Saigal, G, Rodriguez, MM, Podda, A. Langerhans’ cell histiocytosis: pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005; 35:103-115.
- Cochrane, LA, Prince, M, Clarke, K. Langerhans’ cell histiocytosis in the pediatric population: Presentation and treatment of the head and neck manifestation. The Journal of Otolaryngology. 2003; 32(1):33-37.
- Haroche, J, Cohen-Aubart, F, Emile, JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013; 121:1495-1500.
Citation
M G, K S, C S, R K, D A, T A, A T, R T. Langerhans cell histiocytosis. Appl Radiol. 2019;(3):39-41.
June 6, 2019