Ebstein Anomaly
Images
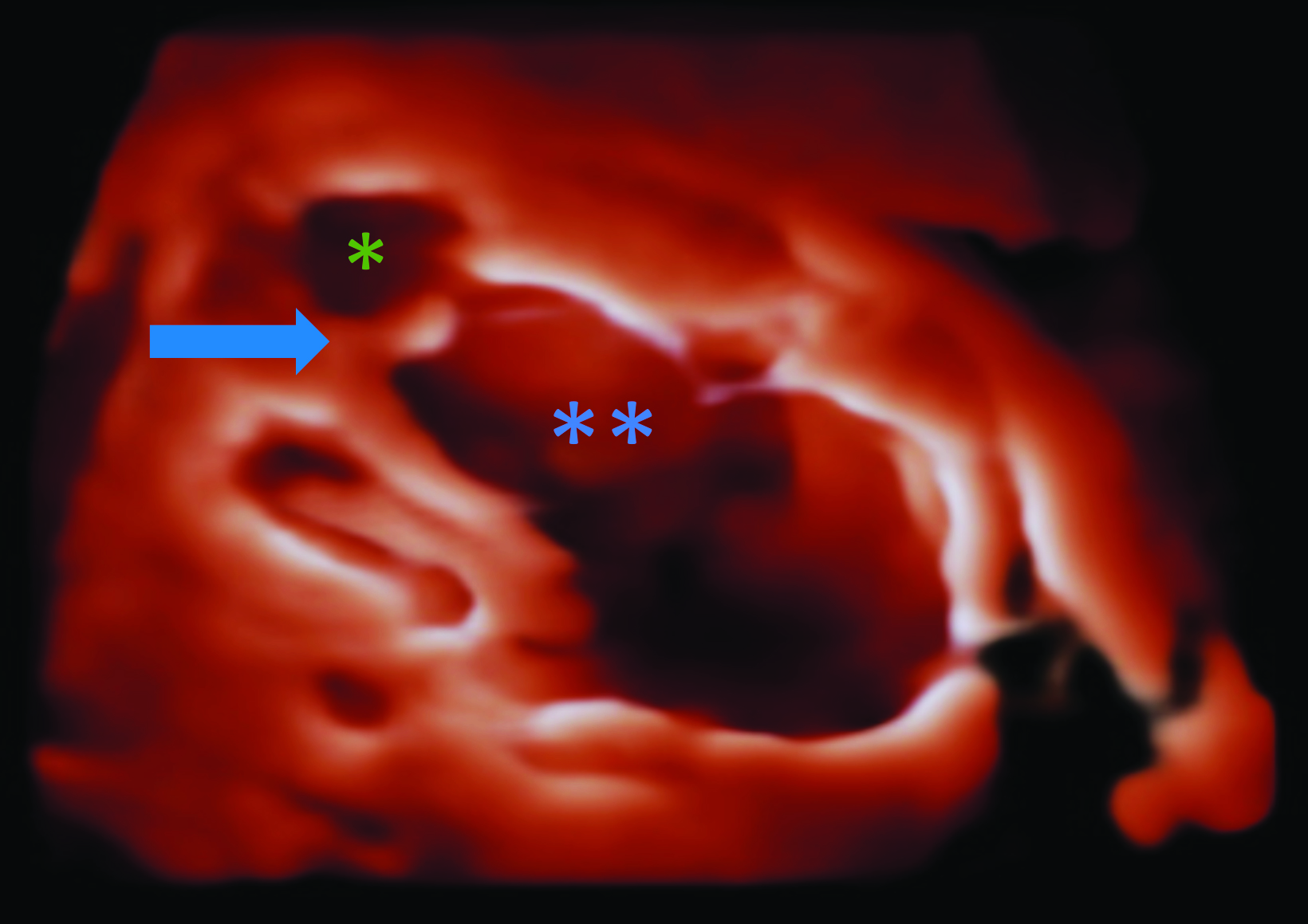
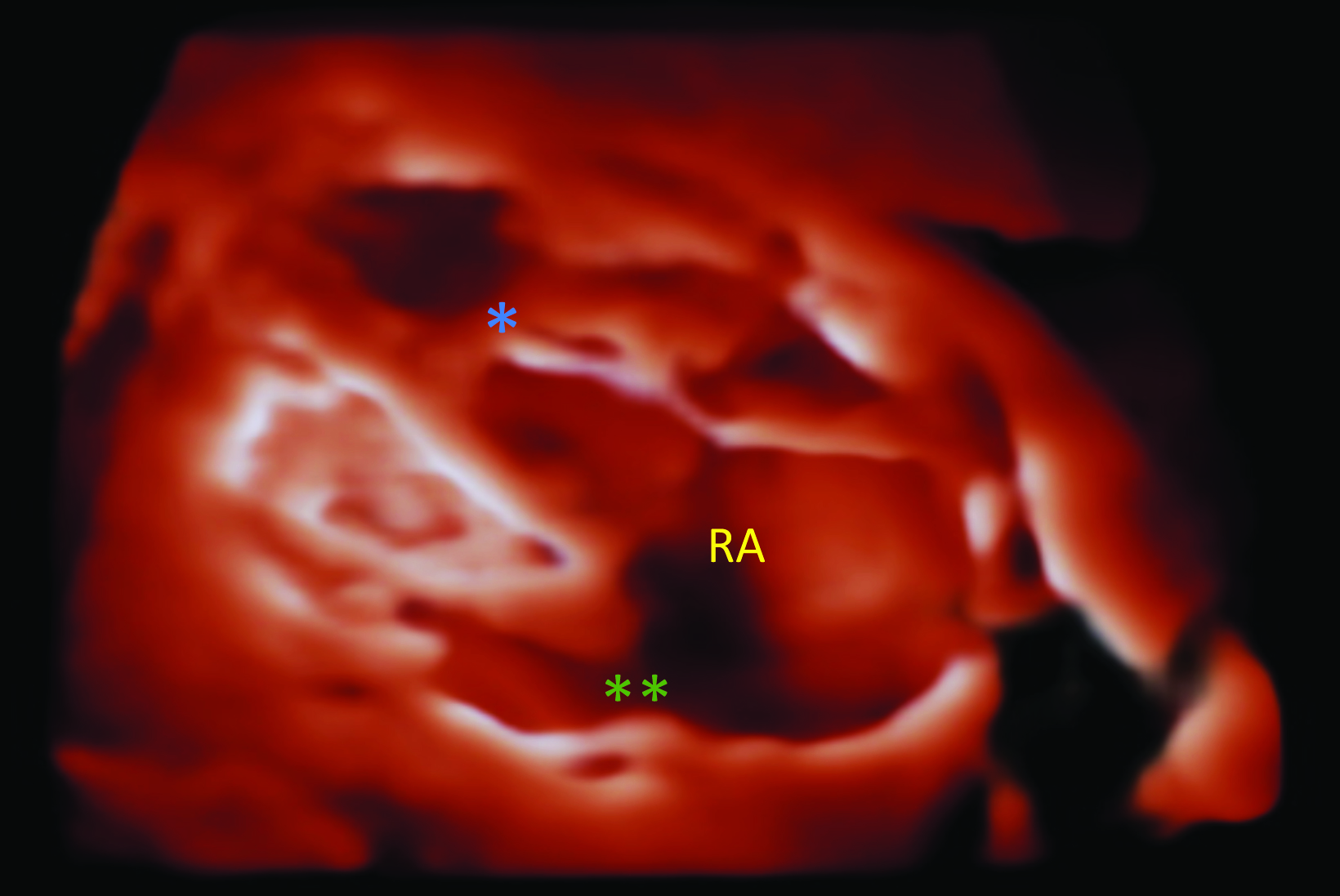
Case Summary
A pregnant patient with a previous infant with Ebstein anomaly (EA) and Trisomy 21 underwent a fetal echocardiogram. The current fetus’s echocardiogram at 25 weeks’ gestation demonstrated apical displacement of the tricuspid valve and a hypoplastic right ventricle.
Imaging Findings
Fetal MRI at 32 weeks 3 days’ gestation demonstrates a markedly enlarged right atrium and atrialized right ventricle. Fetal 4D echocardiogram (Figure) on the same day demonstrated apical displacement of the septal leaflet of the tricuspid valve, marked dilatation of the right atrium, and atrialization of the right ventricle.
Diagnosis
Ebstein anomaly.
The differential diagnosis includes Uhl anomaly, tricuspid valve dysplasia or prolapse, pulmonary valve stenosis, Tetralogy of Fallot, and arrhythmogenic right ventricular cardiomyopathy.
Discussion
Ebstein anomaly is a congenital, right-sided cardiomyopathy characterized by tricuspid valve displacement inferiorly into the right ventricle. It is a rare condition, accounting for 1% of all congenital heart disease.1 While most cases are sporadic, EA has been linked to isolated genetic defects and prenatal lithium exposure.2
Ebstein anomaly is caused by incomplete separation of the septal and posterior leaflets of the tricuspid valve from the underlying myocardium during development. Because the valves fail to separate, they remain attached to the myocardium and are displaced downward, resulting in “atrialization” of the right ventricle.
The atrialized portion of the right ventricle is above the septal and posterior tricuspid valve leaflets and consists of the inlet component of the malformation, which is dilated and contiguous with the right atrium. This ultimately leads to a reduced ventricular volume, decreased right ventricular function, tricuspid regurgitation, right atrial dilation, cardiomegaly, and right heart failure.3
In addition to being regurgitant, the tricuspid valve also has abnormal morphology. The anterior tricuspid leaflet bulges in a sail-like configuration, obstructing right ventricular outflow. This right ventricular outflow obstruction leads to right-to-left shunting, often through a patent foramen ovale or atrial septal defect.4
The clinical presentation of EA varies, depending on the severity of the abnormality, and ranges from asymptomatic with mild atrial displacement to heart failure and death. Features that prompt evaluation for EA include hydrops fetalis and tachyarrhythmias on prenatal ultrasound, cyanosis or heart murmur in neonates and children, and arrythmias or signs of right heart failure in adults.3
Ebstein anomaly is often diagnosed in utero. Routine prenatal ultrasound may show cardiomegaly, tachyarrhythmias, or hydrops fetalis.5 After birth, chest radiograph is often the initial imaging study performed, demonstrating cardiomegaly with a “box-shaped heart” resulting from right atrial enlargement. Ebstein anomaly is also the most common cause of massive cardiomegaly, giving the heart a “wall-to-wall” appearance, touching both the right and left lateral chest walls.
After birth, EA is diagnosed via echocardiography or, if inconclusive, cardiac MRI. Findings include apical displacement of the septal leaflet of the tricuspid valve and an elongated, sail-like anterior tricuspid valve leaflet.3 Echocardiography can determine the severity of EA via the extent of tricuspid regurgitation, through measurements of regurgitant flow velocity and jet area.
The Great Ormond Street Echo (GOSE) score provides prognostic information by predicting the risk of death. The GOSE score is calculated by determining the ratio of the area of the right atrium and atrialized right ventricle to the area of the remaining three cardiac chambers on a standard four-chamber view.6 A GOSE score ≥ 1.5 is uniformly fatal, while scores < 1 have an 8% risk of mortality.7 Cardiac MRI can offer better quantification of EA and has been used to confirm the diagnosis and to grade severity. In today’s practice it is used prior to surgical intervention.8
Once the condition is diagnosed, patients should typically undergo monitoring every 1-2 years with exercise tolerance testing, an electrocardiogram, and cardiac MRI to assess for new signs and symptoms, arrythmias, and progression of cardiac dysfunction.9 Surgical repair is often performed in symptomatic children and adults. Options include tricuspid valve replacement or valvuloplasty, atrialized right ventricle plication, right reduction arterioplasty, and any necessary corrections of arrhythmias or cardiac shunts.10
Prognosis varies with patient age and EA severity. When the condition is diagnosed during the prenatal or neonatal period, prognosis is typically poor with a 30% 1-year mortality rate. Neonates who are cyanotic have a worse prognosis. For example, acyanotic patients with a GOSE Score ratio of 1.1-1.49 have a 10% early mortality rate. This differs from cyanotic patients with the same GOSE Score ratio who have a 100% mortality rate.7 Patients with less-severe disease may be diagnosed in adolescence or early adulthood. These patients have a 20% risk of death by 30 years of age.1
Conclusion
Ebstein anomaly is a rare, congenital, right-sided cardiomyopathy that is characterized by tricuspid valve displacement inferiorly into the right ventricle, resulting in tricuspid regurgitation and eventual right heart failure. The etiology is multifactorial, and most cases are sporadic, with prenatal lithium exposure and isolated genetic defects also being implicated.
Diagnosis is confirmed on echocardiography or cardiac MRI. Ebstein anomaly is managed medically until surgical repair can be performed, and the prognosis is dependent on patient age and symptoms.
References
Citation
MA S, RB T, CM S, AJ T. Ebstein Anomaly. Appl Radiol. 2024;(3):12-13.
May 7, 2024