Duchenne Muscular Dystrophy
Images
Case Summary
An adolescent with a known history of Duchenne muscular dystrophy (DMD) presented for a follow-up neurology evaluation after magnetic resonance imaging (MRI) to evaluate his musculature for fatty infiltration. He was taking glucocorticoids, bisphosphonates, and angiotensin-converting enzyme (ACE)-inhibitors. Physical exam showed impaired neuromuscular function, characterized by abnormal gait, climbing steps with two hands for support, inability to hop on one foot, and height shorter than 1 percentile for age.
Imaging Findings
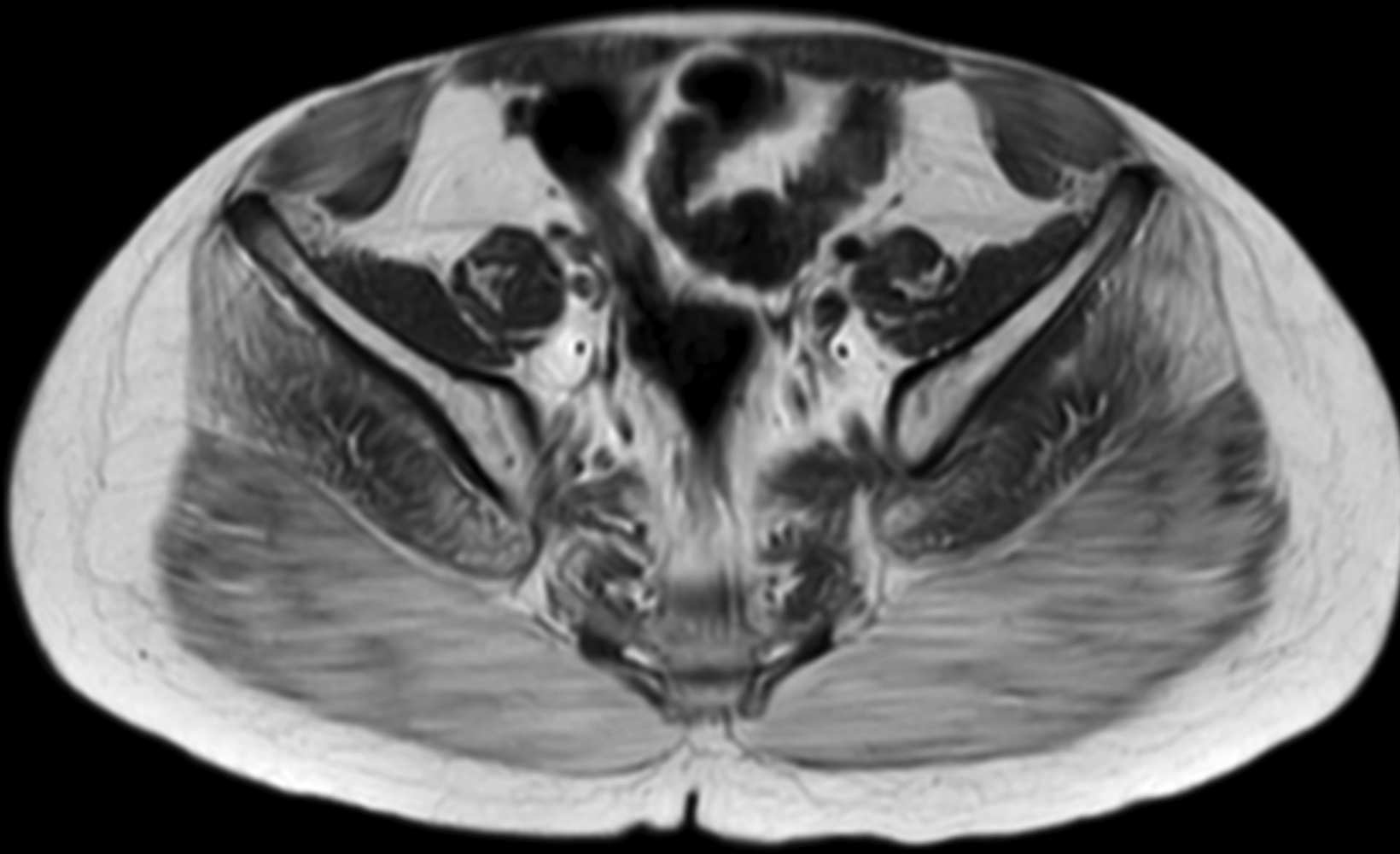
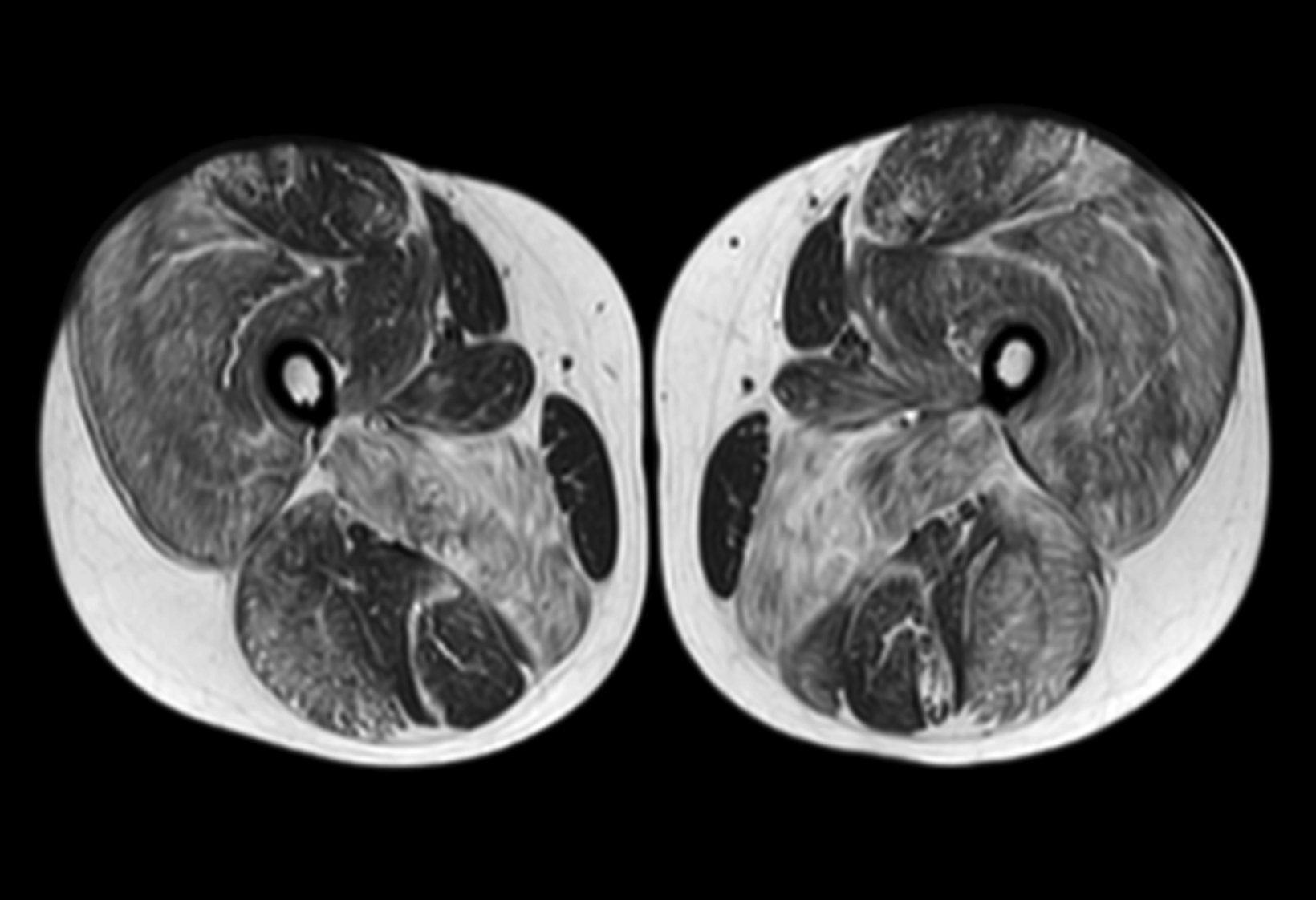
The MRI of the lower extremities revealed fatty infiltration in the musculature of the pelvis and thighs. Notably, there was marked symmetric fatty infiltration of the gluteus maximus muscles, gluteus medius, and adductor magnus muscles. Moderate symmetric fatty infiltration was also seen in the biceps femoris; vastus lateralis, intermedius, and medialis; rectus femoris; and semimembranosus muscles, as well as in the short hip rotators.
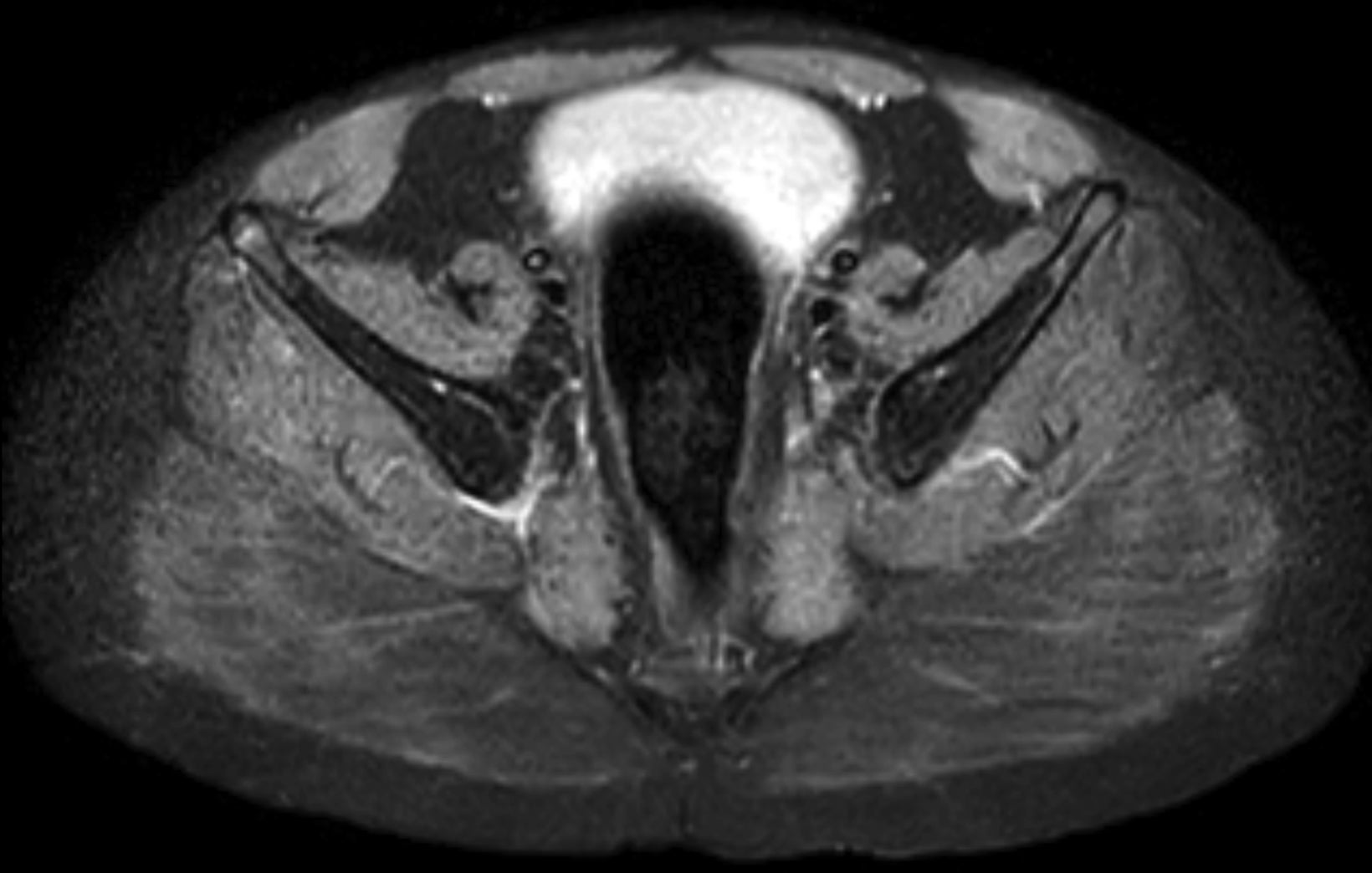
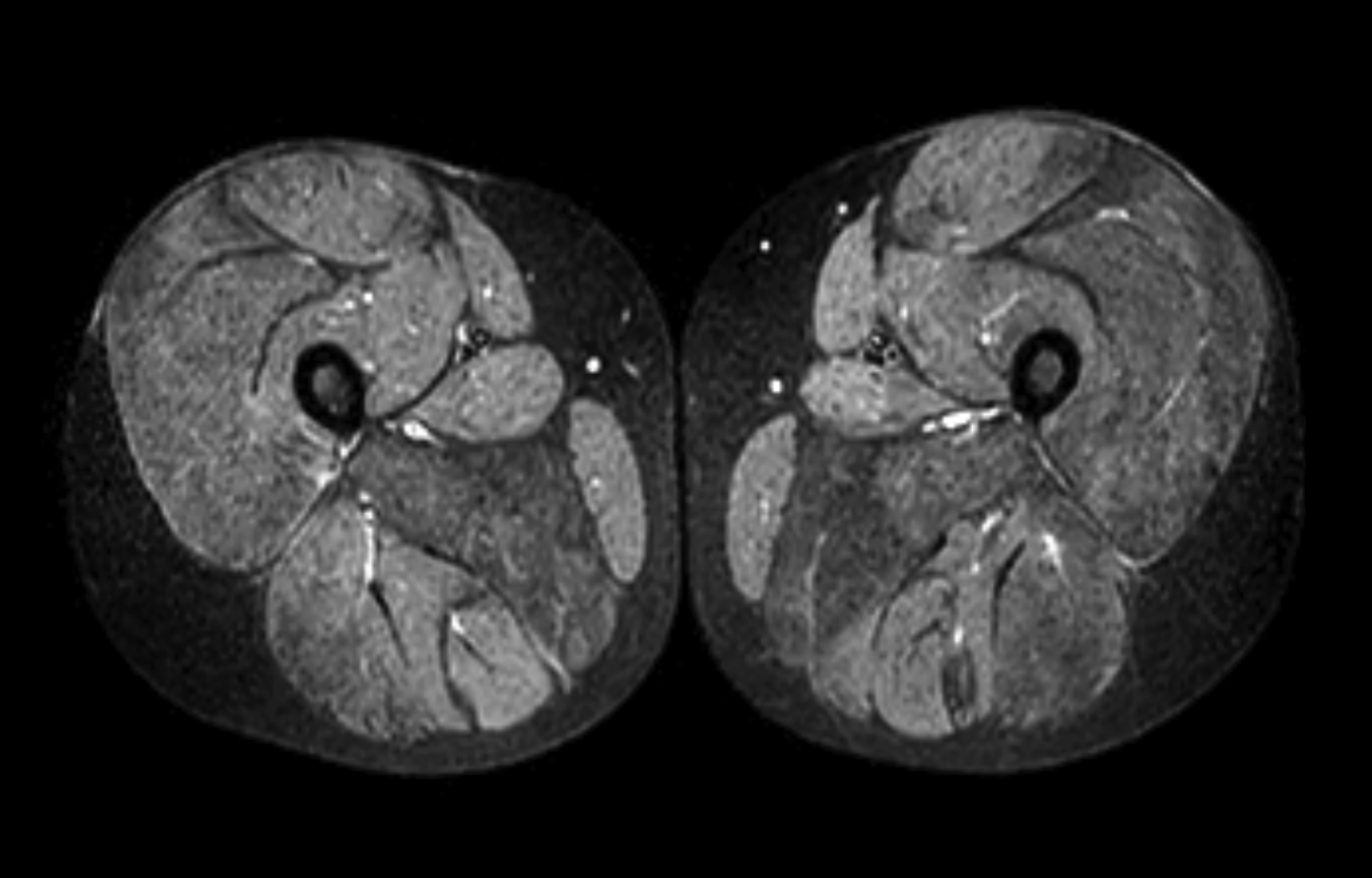
Relative sparing was seen of the sartorius, gracilis, semitendinosus, gluteus minimus, iliacus, and psoas muscles. Mild edema was scattered within several muscles, demonstrated as heterogeneous, mildly increased T2 signal in the musculature around both hips and in the proximal thighs, most notably in the right gluteus medius, left sartorius, and left adductor longus muscles. Coxa valga was present bilaterally.
Diagnosis
Duchenne muscular dystrophy.
Other entities that can look similar on MRI include: Becker muscular dystrophy, limb-girdle muscular dystrophy, and Emery-Dreifuss muscular dystrophy. Additional conditions causing symmetric muscular abnormalities include dermatomyositis, polymyositis, and other longstanding neurologic processes such as cerebral palsy.
Discussion
Duchenne muscular dystrophy is a progressive, X-linked, muscle-wasting disease that results in difficulties with movement and muscle weakness. DMD is caused by a defect in the DMD gene encoding dystrophin, an essential protein in the structural framework of muscle cells.1 Mutations in DMD can also result in Becker muscular dystrophy, a less severe disease with later onset and slower progression. DMD is a rare condition, with prevalence ranging from 1.3 to 2.1 per 10,000 live male births.2
DMD is typically diagnosed in early childhood, with young males presenting with signs of delayed motor milestones, including the inability to walk by age 16 to 18 months, calf pseudohypertrophy, and Gowers sign — the need to use the upper extremities to “walk” up the lower limbs as a way to compensate for weakness in the upper leg and hip muscles.3
Clinical features prompt evaluation of the creatine kinase level, and if it is elevated, genetic analysis is performed to confirm the diagnosis. Other conditions considered in the differential diagnosis of DMD include limb-girdle muscular dystrophy, Emery-Dreifuss muscular dystrophy spinal muscular atrophy, and dilated cardiomyopathy. Genetic analysis is almost always sufficient to confirm diagnosis, but in rare instances a muscle biopsy and immunohistochemistry are necessary.
The pathophysiology of DMD involves dysfunctional dystrophin-mediated sarcolemma weakening, functional ischemia, and free radical damage leading to damaged muscle that is progressively replaced by fat and fibrotic tissue.4 This manifests as varying degrees of muscle weakness, growth delay, cardiomyopathy, scoliosis, and increased incidence of fractures.
While there is no cure for DMD, disease progression is managed with glucocorticoids, typically starting at age 4, followed by serial monitoring of respiratory vital capacity and later cardioprotective ACE-inhibitors.5 Patients often require the use of a wheelchair by age 13. With optimal management most patients have a life expectancy between 20 and 40 years before cardiac and/or respi- ratory failure.6
Important MRI findings include fatty infiltration of muscles, with more than 90% of patients showing moderate-to-severe atrophy of the gluteus maximus, medius, semimembranosus and the vasti.7 The degree of fatty infiltration can be quantified via the modified Mercuri scale,8 from normal muscle appearance (0 points) to end-stage appearance with the entirety of muscle replaced by connective tissue and fat (4 points). Other imaging used in DMD patients may include annual spine dual energy x-ray absorptiometry scanning to measure bone mineral density and echocardiography and/or cardiac MRI to monitor for cardiomyopathy.
MRI has potential in DMD as a biomarker for clinical trials, where some studies have examined MRI correlation with functional outcome measures and its ability to inform treatment response but a robust, quantitative trial is needed.9
Conclusion
Duchenne muscular dystrophy is a progressive, X-linked, muscle-wasting disease diagnosed in early childhood that results in difficulties with movement and muscle weakness. A dysfunctional protein involved in muscle cell structure, dystrophin, leads to muscle damage and eventual replacement by fatty infiltration, which can be monitored and quantified via MRI.
References
Citation
MA S, RB T, CM S, Y L, AJ T.Duchenne Muscular Dystrophy. Appl Radiol. 2023; (2):33-35.
March 7, 2023