Denys-Drash Syndrome
Images
Case Summary
A newborn presented with ambiguous genitalia. Pelvic ultrasound demonstrated no Mullerian structures. Laboratory examination was essentially normal. Genetic testing showed a WT1 variant (a nonsense mutation [c.1999C>T]). Physical examination at 5 months revealed gonads palpable in the inguinal canal, a micro-penis, and hypospadias.
Interval follow-up documented the development of moderate to severe proteinuria, as well as bilateral renal masses. Owing to the patient’s size and young age, with initially small lesions, indeterminate between nephrogenic rests and Wilms tumor, the patient was monitored clinically and via imaging. Ultimately chemotherapy was necessary for obvious enlarging Wilms tumors. One such tumor grew on chemotherapy, prompting expedited surgery. At 10 months, the patient underwent a left heminephrectomy and right radical nephroureterectomy with associated lymph node resection.
Imaging Findings
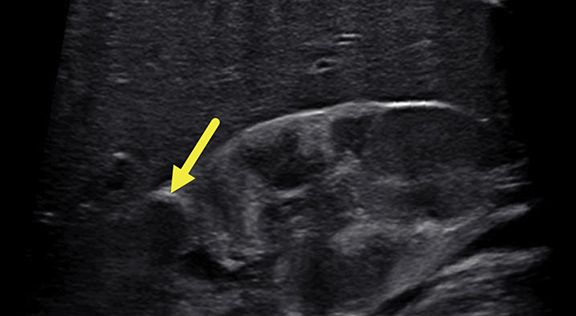
Renal ultrasound at 5 months (Figure 1) revealed enlarged kidneys, above the 95th percentile in length for age. The kidneys were heterogeneous. There was a 1.9 cm, slightly pedunculated, hypoechoic mass in the upper pole of the right kidney and four focal hypoechoic foci in the lower pole. No lesions were found in the left kidney.
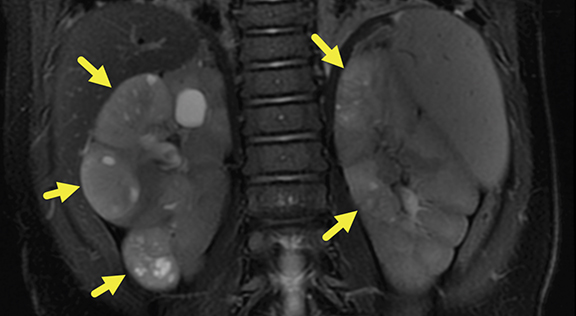
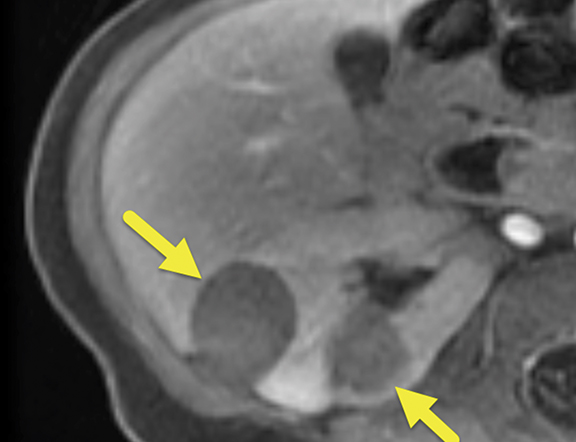
MRI of the abdomen was performed at 6 months (Figure 2), revealing five heterogeneous masses with increased T2 signal, small cystic spaces, and reduced enhancement relative to the renal parenchyma. Three of the masses were present in the right kidney and two in the left. The masses measured 22.6-30.7 mm on the right and 16.8-19.6 mm on the left. Small retroperitoneal and mesenteric lymph nodes were present, none larger than 5 mm in the short axis.
Diagnosis
Denys-Drash syndrome with associated multifocal bilateral Wilms tumors.
There are more than 50 different Wilms tumor predisposition syndromes, including but not limited to Beckwith Wiedemann syndrome and other WT1-mutation associated conditions (WAGR [Wilms tumor, aniridia, genital anomalies, retardation] syndrome and Frasier syndrome).
Discussion
Originally described in 1967 and 1970, Denys-Drash syndrome (DDS) is a rare genetic disorder largely characterized by a triad of gonadal dysgenesis, progressive nephrotic disease, and predisposition for developing Wilms tumor.1,2 DDS symptoms typically present at birth, with kidney disease manifesting within the first few months of life and eventually progressing to end-stage renal disease.3 The inciting genetic mutation is located on chromosome 11p13 at the Wilms tumor suppressor gene (WT1). Mutation in the WT1 gene leads to disruption of regular kidney and gonad development.
In DDS, the majority of affected individuals have heterozygous germline missense pathogenic mutations in exons 8 or 9, resulting in an inability for the WT1 protein to properly bind DNA.4,5 DDS has an autosomal dominant pattern, with most cases occurring in individuals with no family history and stemming from de novo mutations in the WT1 gene.3,5 The incidence and prevalence of DDS is currently unknown and shows no difference among race or ethnicity.
Most children with DDS present with ambiguous genitalia. However, there are cases where newborns display typical male external genitalia or genitalia that appear phenotypically female. While both sexes are affected by DDS, the relative lack of female cases may be due to delayed or missed diagnosis.3,6 Internal genitalia have been found to be extremely variable in DDS, with various cases demonstrating Wolffian structures, both Mullerian and Wolffian structures, dysgenic gonads, or the presence of both testicular and ovarian tissue. Previously, cases have shown the development of gonadoblastoma in DDS, yet the risk remains low compared to other conditions with mutations in WT1 such as Frasier Syndrome.3
Typically at birth or within the first year of life children present with a primary and progressive nephropathy.1-2,6-7 The salient histological feature on renal biopsy is diffuse mesangial sclerosis along with thickened glomerular capillary basement membranes, and tubulointerstitial inflammation. This nephropathy is generally steroid resistant and associated with severe hypertension and proteinuria. In DDS, the nephropathy typically progresses within the first two to three years to end-stage renal disease and renal failure. The cumulative incidence of end-stage renal disease at 20 years from diagnosis was found to be 74% in DDS.7 Consequently, the major cause of death among DDS patients is renal failure secondary to the nephropathy.
The third component of the DDS triad is a strong predilection for development of Wilms tumor. In DDS, the median age for Wilms tumor presentation was found to be 12.5 months, commonly as a palpable abdominal mass.5 Not every case of DDS has reported Wilms tumor, however. The actual risk of Wilms tumor development may be skewed, as patients may undergo renal transplant or die from renal failure before cancer development.3,5 Additionally, the clinical presentation of DDS includes a constellation of other symptoms, including: oliguria, edema, decreased activity growth delay, abdominal distention/pain, hematuria, and weight loss.
Management of DDS is multidisciplinary and may include a team represented by nephrology, oncology, surgery, urology, endocrinology, radiology, and genetics.8 Medical care is focused on treatment and monitoring of the progressive nephropathy and subsequent end-stage renal disease. This largely includes managing electrolyte and fluid balance, treating hypertension, and dialysis.8
Decisions over dialysis method (hemodialysis or peritoneal dialysis) depend on other factors related to care, yet peritoneal dialysis may be preferable for infants based on their size and therapy tolerance. Moreover, standard chemotherapy for treatment of DDS-associated Wilms tumor includes vincristine, actinomycin D, and sometimes doxorubicin, as Wilms tumors associated with DDS are invariably of the favorable histology subtype.
Two major approaches to treatment have been described in the literature: an aggressive approach of bilateral pre-emptive nephrectomy with shortened dialysis time prior to renal transplantation, and a conservative approach with frequent imaging to delay dialysis and transplantation, which is less technically difficult with increasing age. Gariepy-Assal, et al, suggest the preservation of renal function as the guiding principle, and that bilateral nephrectomy should be performed once end-stage renal disease occurs.9
Overall, treatment should be individualized and factor in the progression of renal failure, the age of the child, the amenability to nephron-sparing approaches when tumors arise, and hypertension control. Regular imaging is necessary for proper surveillance of Wilms tumor development, progression, and/or recurrence. Pelvic and abdominal ultrasonography are indicated in patients presenting with symptoms suggesting DDS to evaluate for the presence of Wilms tumor and the presence of abnormal internal genitalia.8 Abnormalities indicate use of pelvic and abdominal CT or MRI, as these modalities are more sensitive in evaluating Wilms tumors.
Unfortunately, in this case, while one-half of one kidney was spared, the remnant kidney, with ongoing nephrotic range proteinuria, quickly decompensated to end-stage renal disease. The patient completed postoperative radiotherapy indicated by local stage of cancer progressed to postoperative chemotherapy, on hemodialysis. Renal transplantation will ultimately be considered following a period of observation and confirmation of a sustainable remission.
Conclusions
Denys-Drash syndrome is a rare condition affecting infants and children and characterized by a triad of ambiguous genitalia, nephrotic syndrome pathognomonic of diffuse mesangial sclerosis, and predisposition for Wilms tumor development. Genetic testing indicates a mutation at the WTI gene located on chromosome 11p13, resulting in an abnormal WTI protein. Renal ultrasound is useful in screening for Wilms tumors and abnormal internal genitalia. Abdominopelvic CT or MRI is important for monitoring renal tumors. DDS management is highly individualized and focuses on preserving renal function and balancing the risk of end-stage renal disease progression and Wilms tumor progression.
References
- Denys P, Malvaux P, Van Den Berghe H, Tanghe W, Proesmans W. Association of an Anatomo-Pathological Syndrome of Male Pseudohermaphroditism, Wilms’ Tumor, Parenchymatous Nephropathy and XX/XY Mosaicism. Arch Fr. Pediatr. 1967; 24(7): 729-739.
- Drash A, Sherman F, Hartmann WH, Blizzard RM. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatrics. 1970; 76(4): 585-593.
- Meuller RF. The Denys-Drash Syndrome. J Med Gen. 1994; 31: 471-477.
- Dome JS, Huff V. Wilms Tumor Predisposition. 2003 Dec 19 [Updated 2016 Oct 20]. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1294/.
- Roca N, Muñoz M, Cruz A, Vilalta R, Lara E, Ariceta G. Long-term outcome in a case series of Denys-Drash syndrome. Clinical Kidney Journal. 2019; 12(6): 836-839.>Eddy AA, Mauer M. Pseudohermaphroditism, glomerulopathy, and Wilms tumor (Drash syndrome): Frequency in end-stage renal failure. J Pediatr. 1985; 106(4): 584-587.
- Breslow N. End Stage Renal Disease in Patients with Wilms Tumor: Results from the National Wilms Tumor Study Group and the U.S. Renal Data System. J Urol. 2005; 174(5): 1972-1975.
- Auber F, Jeanpierre C, Denamur E, Jaubert F, Schleiermacher G, Patte C, Cabrol S, Leverger G, Nihoul-Fékété C, Sarnack S. Management of Wilms tumors in Drash and Frasier syndromes. Pediatr Blood Cancer. 2009; 52(1): 55-59.
- Gariépy-Assal L, Gilbert RD, Žiaugra A, Joy Foster B. Management of Denys-Drash syndrome: A case series based on an international survey. Clin Nephrol.Case Studies. 2018; 6: 36-44.
References
Citation
SS G, RB T, Y L, JI G, AJ T,. Denys-Drash Syndrome. Appl Radiol. 2021;(5):61-63.
September 10, 2021