Cystic Biliary Atresia
Case Summary
An infant born at 33 weeks’ gestation presented with neonatal cholestasis. Total and direct bilirubin levels were significantly elevated. The patient’s stool color was normal. Mild jaundice and scleral icterus were found on examination.
Imaging Findings
Ultrasound (US, Figure 1 ) showed a 1 mm × 8-mm atretic gallbladder, despite the infant having nothing by mouth for 5 hours prior to the study. There were no visible intrahepatic bile ducts. Additionally, a 5 × 4-mm cyst was present near the hepatic hilum.
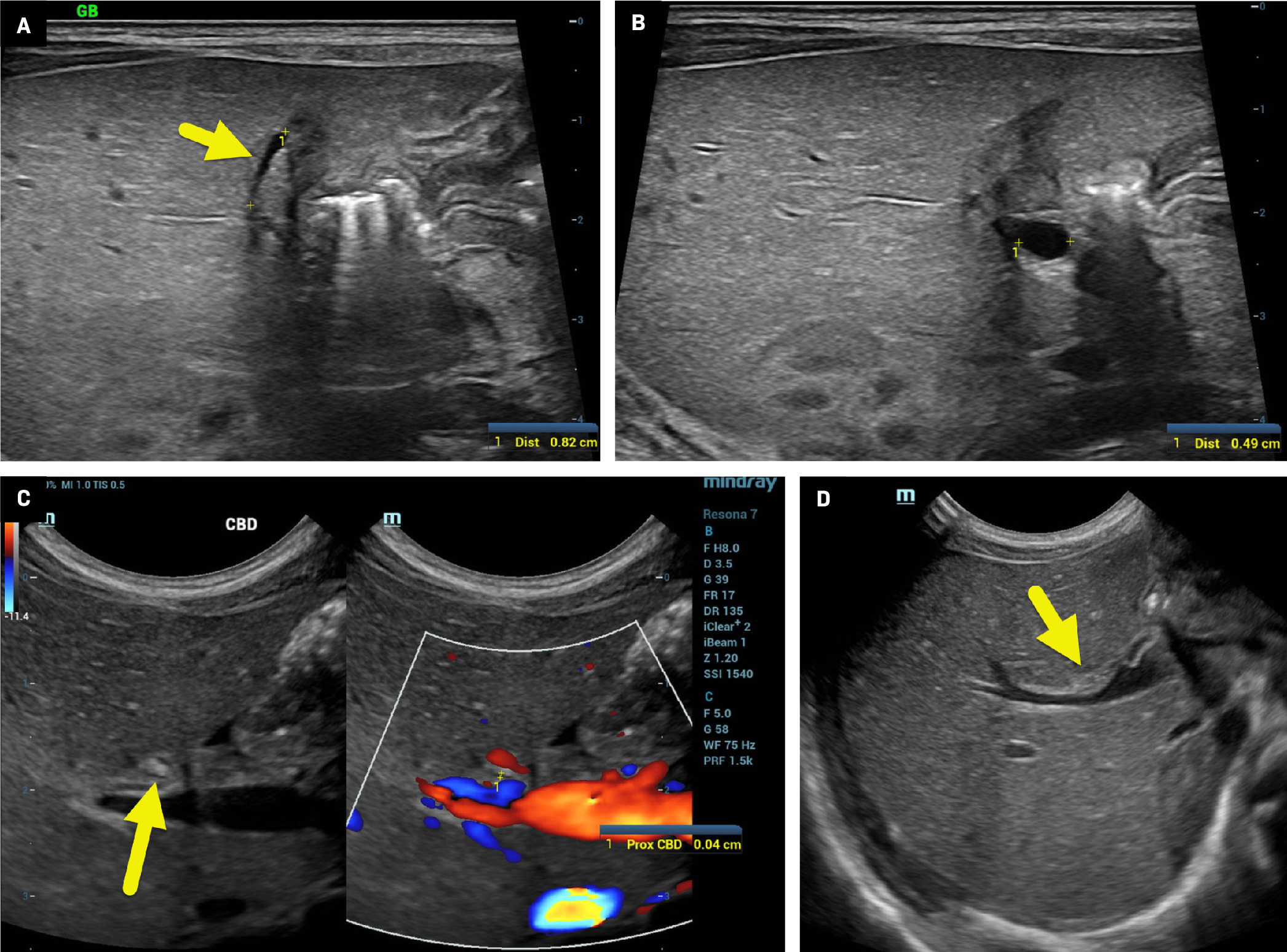
US of the liver showed an atretic gallbladder (A, arrow) 8 mm in length and 1 mm in diameter. A 5 mm cyst (B, calipers) was present at the hepatic hilum. The common bile duct (C) was not definitively identified. However, a candidate duct (arrow) was 0.4 mm in diameter. There was a small triangular cord sign (D, arrow), 2 mm in diameter.
MRI cholangiopancreatography (MRCP) ( Figure 2 ) confirmed gallbladder atresia and the cyst in the hepatic hilum. The common bile duct was not visible.
Coronal imaging (A) from MRCP and axial (B) and coronal T2-weighted images (C) showed a small cyst (arrow) at the hepatic hilum. There were no visible gallbladder or intrahepatic bile ducts. Axial T2-weighted imaging (D) near the portal bifurcation (arrow) showed increased signal related to inflammation, with no visible bile ducts. Note the diffuse, low T2 signal throughout the liver.

Intraoperative cholangiogram ( Figure 3 ) showed filling of the cystic structure but no contrast within intrahepatic bile ducts or draining into the duodenum.
Intraoperative cholangiogram showed contrast injected directly into the cystic structure (arrow). There was no extension of contrast proximally into intrahepatic bile ducts or distally to the duodenum.

Diagnosis
Cystic biliary atresia.
Differential diagnosis includes choledochal cyst and Alagille syndrome.
Discussion
Biliary atresia is a progressive congenital disease involving sclerosing inflammation and obstruction of the intra- and extrahepatic bile ducts.1, 2 While it is the most common cause of cirrhosis and hepatic failure in children, its incidence varies by region. BA is most common in Taiwan, where it affects 1 in 5000 live births, and least common in the Netherlands, where it affects 1 in 19,000 live births.2 - 4 Patients typically present with jaundice, pale-colored stool, dark urine, coagulopathy, and failure to thrive. Chronic findings include cirrhosis, hepatosplenomegaly, and ascites.3
BA is classified according to the most proximal level of the biliary obstruction. In type 1 BA (~5% of patients), the biliary system is patent to the level of the proximal common bile duct at the cystic duct insertion. There is atresia of distal common bile duct. In type 2 BA (~2% of patients), the common hepatic duct is atretic, but intrahepatic ducts are patent and cystic ducts can be found at the porta hepatis. In type 2A, the common bile duct is patent while in type 2B the cystic ducts and hepatic ducts are not patent. Finally, in type 3 BA (~90% of patients), the central intrahepatic ducts and the extrahepatic biliary are atretic.1, 3 Cystic biliary atresia (CBA) is an uncommon variant of BA accounting for 5% to 10% of patients with BA.1 CBA is characterized by bile duct atresia, with cystic changes in the common hepatic or common bile duct.1
Abdominal US is the initial imaging modality in patients with suspected BA. In children younger than 1 year, a healthy gallbladder should measure 15 to 30 mm in length and should have thin, defined walls.5 Patients with BA may have findings known as the gallbladder “ghost triad,” with a gallbladder less than 19 mm in length with a thin, indistinct wall and an irregular or lobulated contour.5 Nonvisualization of the gallbladder alone on US cannot confirm BA.5 Other than gallbladder abnormalities, additional US findings of BA include triangular cord sign, absence/nonvisibility of the common bile duct, and hepatic artery enlargement.1 - 5 The triangular cord sign represents a triangular or tubular echogenic cord of fibrous tissue more than 4 mm in thickness along the anterior wall of the right portal vein, representing an obliterated bile duct.2, 5 Prior studies have shown that a hepatic artery diameter greater than or equal to 1.5 mm has a high sensitivity, specificity, and accuracy for BA.5
Although abdominal US is the primary imaging modality used to evaluate BA, an MRI, MRCP, nuclear medicine hepatobiliary iminodiacetic acid (HIDA) scan, and cholangiography may also be used for diagnosis. MRI and MRCP are more commonly used to exclude other causes of cholestasis.5 MRI and MRCP findings in patients with BA are similar to US findings. When using MRI, findings for BA are triangular cord thickness greater than or equal to 5.1 mm, nonvisualization of the common bile duct, and abnormal gallbladder.5 During HIDA scans, an injected hepatobiliary agent is taken up by hepatocytes. If the radiotracer does not excrete into the small bowel after 24 hours, a diagnosis of BA can be suggested.5 However, neonatal hepatitis can have similar HIDA scan findings. Percutaneous or intraoperative cholangiogram may also be performed. The findings of BA include failure of contrast to opacify into the intrahepatic and extrahepatic bile ducts .5
BA is ultimately diagnosed via biopsy. An adequate liver biopsy should be at least 2.0 cm long × 0.2 mm wide, and ideally contain at least 10 portal areas.6 In BA, the liver has expanded, edematous myofibroblastic portal tracts with bile duct proliferation and anastomosing ductules at the periphery of the portal tracts.6 Ductular bile plugs, fibroblast proliferation, and inflammatory cells, particularly neutrophils, are characteristic on histopathology.6 In CBA, the cyst has a thin, uniform layer of dense scar tissue with very few cells that tend to peel from the inner cyst surface. There is also focal myofibroblastic hyperplasia enclosing the inner scarred layer.7
BA is treated via the Kasai procedure, which includes hepatic portoenterostomy, biliary remnant resection, and creation of Roux-en-Y intestinal anastomosis for bile flow.1, 6 If the Kasai procedure fails or the patient develops cirrhosis, a liver transplant is required.6 Treatment after 60 postnatal days can accelerate hepatic fibrosis and liver failure, worsening patient outcomes.2, 6 The rapid development of cirrhosis in patients with BA makes it crucial to differentiate CBA from choledochal cyst.
CBA and choledochal cyst have many clinical and imaging similarities, which can make distinguishing between CBA and choledochal cyst challenging. Gallbladder morphology, triangular cord thickness, and cyst size, contents, and progression are used to differentiate CBA from choledochal cyst. A normal-sized gallbladder (> 15 mm) is characteristic of choledochal cyst, whereas an atretic (< 15 mm) or abnormally shaped (tubular or elongated) gallbladder is characteristic of CBA.1, 2, 4 The triangular cord sign on US or MRI and a cyst size under 1.5 cm on prenatal US are diagnostic for CBA.1, 2, 4 Prior studies have also demonstrated that the cyst size in CBA stays stable in the prenatal phase and regresses after birth. Patients with choledochal cyst had continuous growth of cysts after birth.4 When incised, choledochal cysts contain dark-green bile and biliary sludge, whereas cysts in patients with CBA have no content or small amounts of yellow fluid.1, 2, 4
Conclusion
CBA is an uncommon variant of BA. Distinguishing CBA from choledochal cyst is paramount for patients to receive timely treatment and have the best outcomes.
References
Citation
O’Connor BG, Towbin RB, Schaefer CM, Towbin AJ. Cystic Biliary Atresia. Appl Radiol. 2024;(5):3 - 6.
doi:10.37549/AR-D-24-0018
October 1, 2024