Craniopharyngioma
Images

Case Summary
A previously healthy 13-year-old presented with headaches, visual impairment, and new onset of seizures.
Imaging Findings
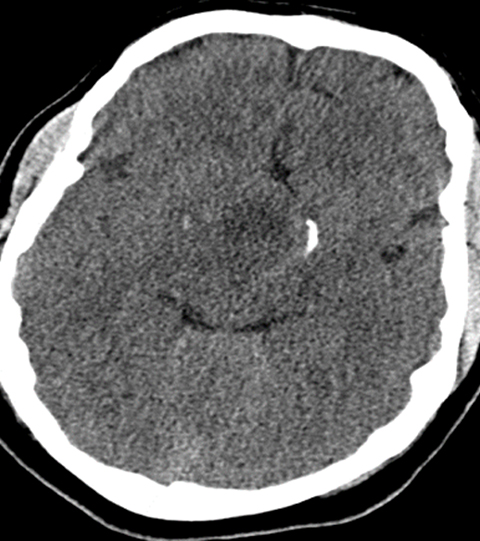
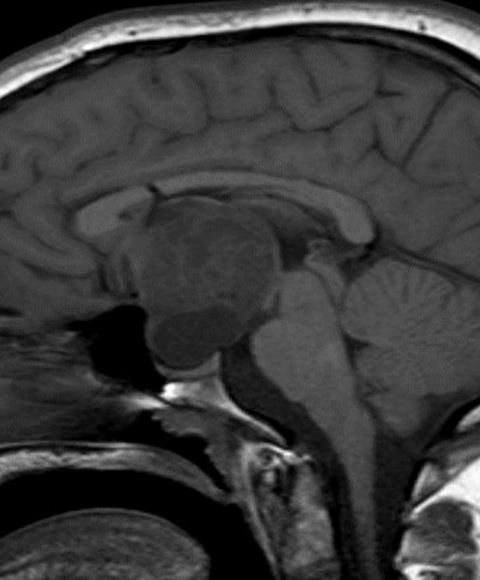
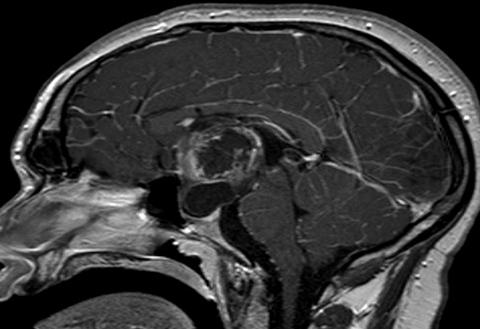
Head CT (Figure 1) demonstrated a large, hypodense, suprasellar mass. The lesion had a large, rounded hypodense component that exerted mass effect on adjacent structures, including the posterior aspect of the anterior interhemispheric fissure and the midbrain. The lesion was bracketed by comma-shaped calcifications laterally. Brain MRI (Figure 2) revealed a large suprasellar mass consisting of cystic and solid components. The solid tissue in the rim of the lesion enhanced after contrast injection.
Diagnosis
Craniopharyngioma. The differential diagnosis of an intrasellar/suprasellar mass in childhood includes craniopharyngioma, Rathke cleft cyst, pituitary macroadenoma, and intracranial teratoma. Langerhans cell histiocytosis, lymphocytic hypophysitis, and sarcoidosis may involve the pituitary infundibulum.
Discussion
Craniopharyngiomas are rare tumors, with an incidence of 0.13-0.18 cases per 100,000 in the US1 and a bimodal age distribution. Peak incidence in children ranges from 6 to 15 years of age, and in adults over 40 years. Craniopharyngiomas account for 6 to 9% of all pediatric brain tumors.1 Given their typical locations within the sella turcica with suprasellar extension, these tumors are especially challenging to resect and can cause significant neurological complications.
Headaches, caused by increased intracranial pressure, are present in 60-75% of patients and are one of the most common symptoms at presentation.2 Visual deficits are the second-most common presenting symptoms, occurring in more than 50% of patients. Children will often report being unable to see the board at school or the television at home.2
Endocrine abnormalities are present in 20 to 50 % of patients and can include diabetes insipidus, obesity, growth retardation, and delayed puberty related to compression and/or invasion of the hypothalamic-pituitary stalk.2 As the tumor grows, it can cause frontal lobe compression and/or obstructive hydrocephalus. Children with large tumors can thus present with cognitive and functional deterioration typically manifested as poor school performance or psychomotor decline.2 Importantly, craniopharyngiomas are slowly growing tumors. Thus, symptom onset can be insidious, leading to delayed diagnosis. Presentation also tends to vary by age — younger children manifest with symptomatology of elevated intracranial pressure, whereas adolescents present more commonly with hypopituitarism and visual deficits.
Imaging is critical in evaluating craniopharyngiomas, specifically with regard to tumor location and adjacent structure involvement. These factors are important in determining the best course of treatment. Tumor involvement of and/or proximity to the frontal and temporal lobes, ventricles, optic nerves and chiasm, hypothalamus, pituitary gland, circle of Willis, and brainstem all impact the treatment plan.2 While MRI is generally accepted as the best imaging modality to visualize crucial information about craniopharyngiomas, CT may be additive.
On CT, the tumor can have heterogeneous density with cystic and solid components. Typically, the cystic components are dominant. Approximately 90% of lesions contain calcifications located peripherally. Secondary skull base changes such as enlargement of the sella turcica or erosion of the dorsum sellae can be seen.3
The MRI appearance may vary depending on the cystic component. On T1-weighted sequences, the cystic components may be isointense to hyperintense to brain. Hyperintensity, when present, results from the high protein content of the cyst fluid. On T2-weighted sequences, the lesion has mixed hyperintense signal. The solid portions of the tumor enhance with injection of contrast.3
Magnetic resonance angiography may be valuable to evaluate tumor proximity to the internal carotid arteries and adjacent portions of the circle of Willis.3 Magnetic resonance spectroscopy (MRS) can be particularly useful in distinguishing craniopharyngiomas from gliomas and pituitary adenomas; however, this can be technically challenging with smaller lesions near the skull base. On MRS, craniopharyngiomas demonstrate characteristic lactate or lipid peaks, with only small amounts of other metabolites, whereas gliomas typically demonstrate choline, n-acetyl aspartate, and creatine peaks, and pituitary adenomas demonstrate choline peaks or no elevated metabolites.4
There are two identified subtypes of craniopharyngioma – adamantinomatous (aCP) and papillary (pCP). The former is the more common of the two. Although they are similar histologically, it has been hypothesized that the subtypes exhibit distinctly different pathogenesis.
Childhood craniopharyngiomas are nearly exclusively aCPs. These are hypothesized to develop from neoplastic transformation of embryonic epithelial remnants of the Rathke pouch or the craniopharyngeal duct, the embryonic origin giving rise to the anterior pituitary as it ascends from the pharynx into the future sella turcica region during embryogenesis.5 In contrast, pCPs are seen almost exclusively in adults, presenting as solid tumors with little mineralization.2 They are thought to result from metaplasia of differentiated pituitary cells.
Craniopharyngiomas are generally benign and rarely undergo malignant transformation.2 Both subtypes are considered by the World Health Organization to be grade 1 tumors. Nevertheless, their proximity to vital neurovascular structures causes symptoms that not only can lower quality of life but can also be fatal. Complete surgical resection can be curative; however, treatment often also incorporates irradiation.
Surgical resection can be via a craniotomy or endoscopically. Given the significant morbidity associated with open approaches, the endoscopic, endonasal/transsphenoidal approach, when feasible, has become more popular. Its advantages include enhanced visualization with panoramic endoscopic view and lack of external incision. In pediatric patients, however, the endoscopic transsphenoidal approach can be more challenging given incomplete development of the sphenoid sinus.
Surgical management, especially in children, remains controversial. The best treatment for craniopharyngioma is the one that involves the least long-term morbidity. Treatment may include surgery only, irradiation only, or, more commonly, a combination of the two. Surgery alone implies radical resection, and is, therefore, appropriate for tumors that may be completely resected without neurovascular injury and visual impairment. Limited surgery and radiation therapy are appropriate for most patients including those for whom radical surgery might be considered. The concept of limited surgery and radiation therapy involves the use of surgery to alleviate and prevent symptoms, improve a patient’s ability to tolerate and complete irradiation, and potentially enhance or optimize dose delivery6.
Conclusion
Craniopharyngioma is the most common non-glial intracranial tumor of childhood. Because of the tumor’s slow growth, the onset of symptoms can be insidious. Patients typically present with headaches, visual field defects, or endocrine anomalies. On imaging, the mass has variable cystic and solid areas. Peripheral calcifications are often present. One of the primary goals of imaging is to determine disease extent and tumor relationship to vital neurovascular structures. While surgical resection and/or radiation therapy can be curative, the central location in the brain of these tumors poses risks of long-term morbidity.
References
- Yang I, Sughrue ME, Rutkowski MJ, Kaur R, Ivan ME, et al. Craniopharyngioma: a comparison of tumor control with various treatment strategies. Neurosurg Focus. 2010; 28(4):E5
- Drapeau A, Walz P, Eide J, Rugino A, Shaikhouni A, et al. Pediatric craniopharyngioma. Child’s Nervous System. 2019;35(11); 2133-2145.
- Schroeder JW, Vezina LG. Pediatric sellar and suprasellar lesions. Pediatr Radiol. 2011; 41(3): 287–298. Quiz 404-405.
- Sutton LN, Wang ZJ, Wehrli SL, Marwaha S, Molloy P, Phillips PC, Zimmerman RA. Proton spectroscopy of suprasellar tumors in pediatric patients. Neurosurgery. 1997; 41(2):388–394; discussion 394-395.
- Larkin, S. and Ansorge, O. Pathology and pathogenesis of craniopharyngiomas. Pituitary. 2012; 16(1): 9-17.
- Müller H, L: The Diagnosis and Treatment of Craniopharyngioma. Neuroendocrinology. 2020;110:753-766. doi: 10.1159/000504512.
Citation
AN Dominianni, Towbin RB, Schaefer CM, Towbin AJ, Aria DJ. Craniopharyngioma. Appl Radiol. 2021;(5):58-60.
September 10, 2021