Craniometaphyseal Dysplasia
Images
Case Summary
A teenager presented with a facial deformity and difficulty breathing through his nose since childhood.
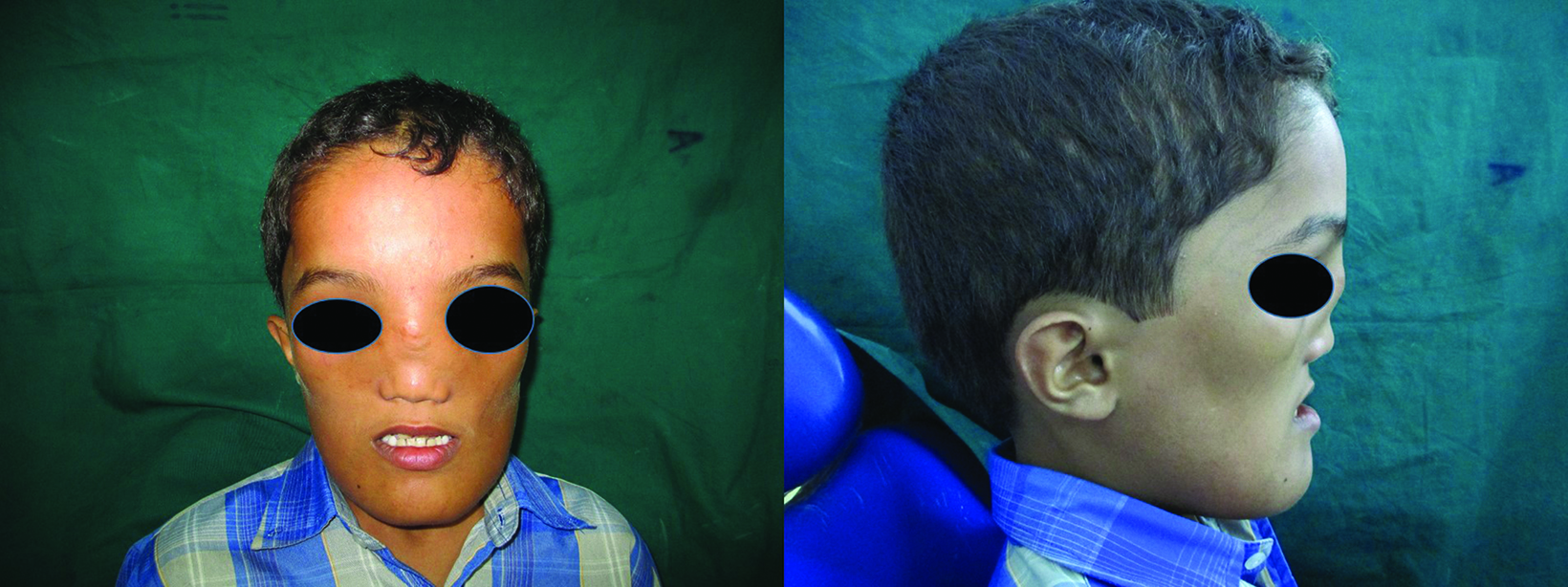
Physical examination revealed hypertelorism, convergent squint, glabellar protrusion, hypoplastic maxilla, midfacial deficiency, depressed nose with broadened root of the nose, and malar prominence.
Prominent frontonasal bone bossing was also evident (Figure 1). The patient had diminished visual acuity, sensorineural hearing loss, and difficulty breathing through the nose. The patient’s mental and speech development were unimpaired, while physical development was mildly delayed. Intraoral examinations showed retained maxillary and mandibular canines and molars, and an absent maxillary left lateral incisor. The mandibular anterior teeth were in separate crossbite and relation to the maxillary anterior teeth with Class III malocclusion.
Imaging Findings
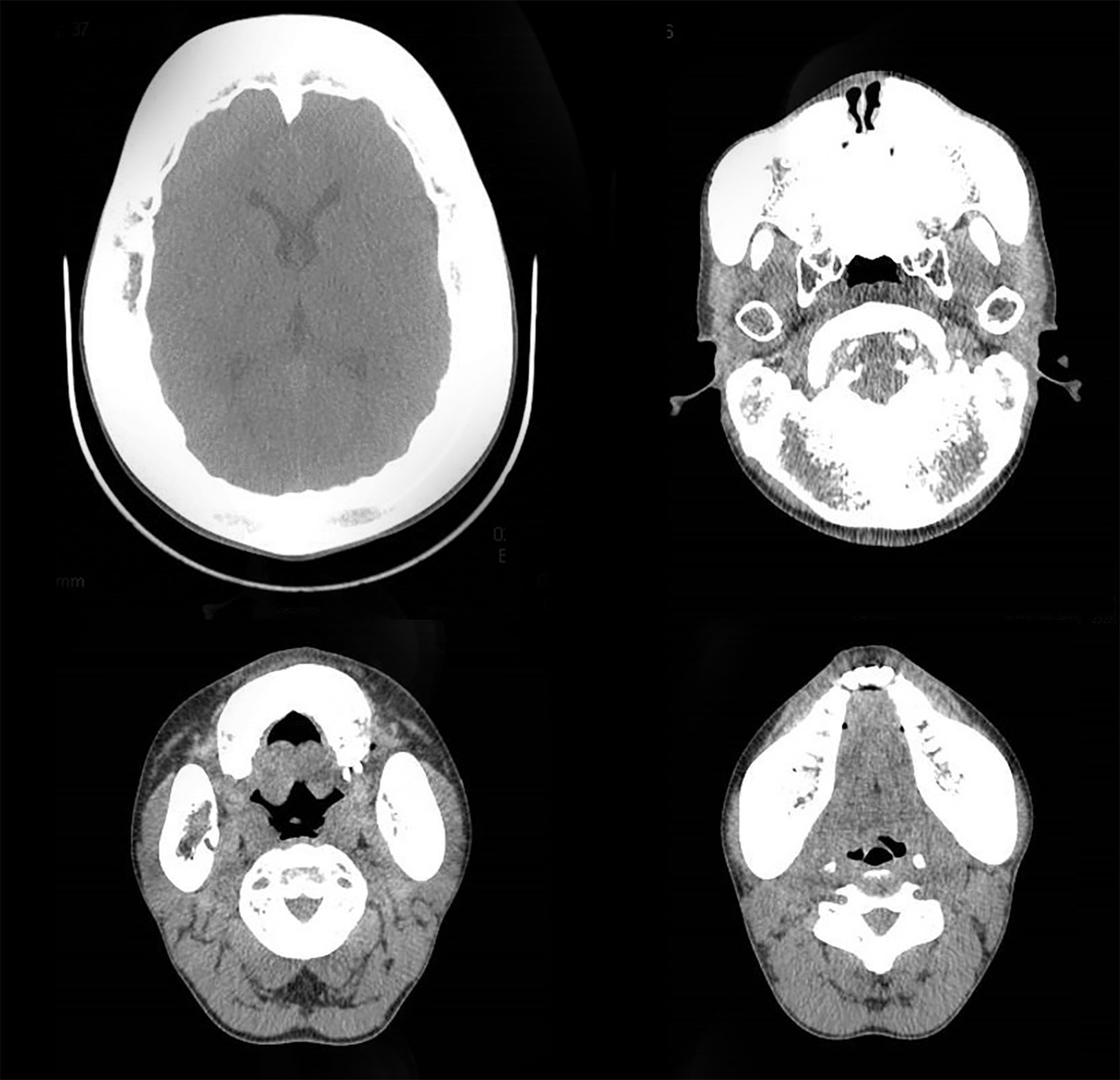
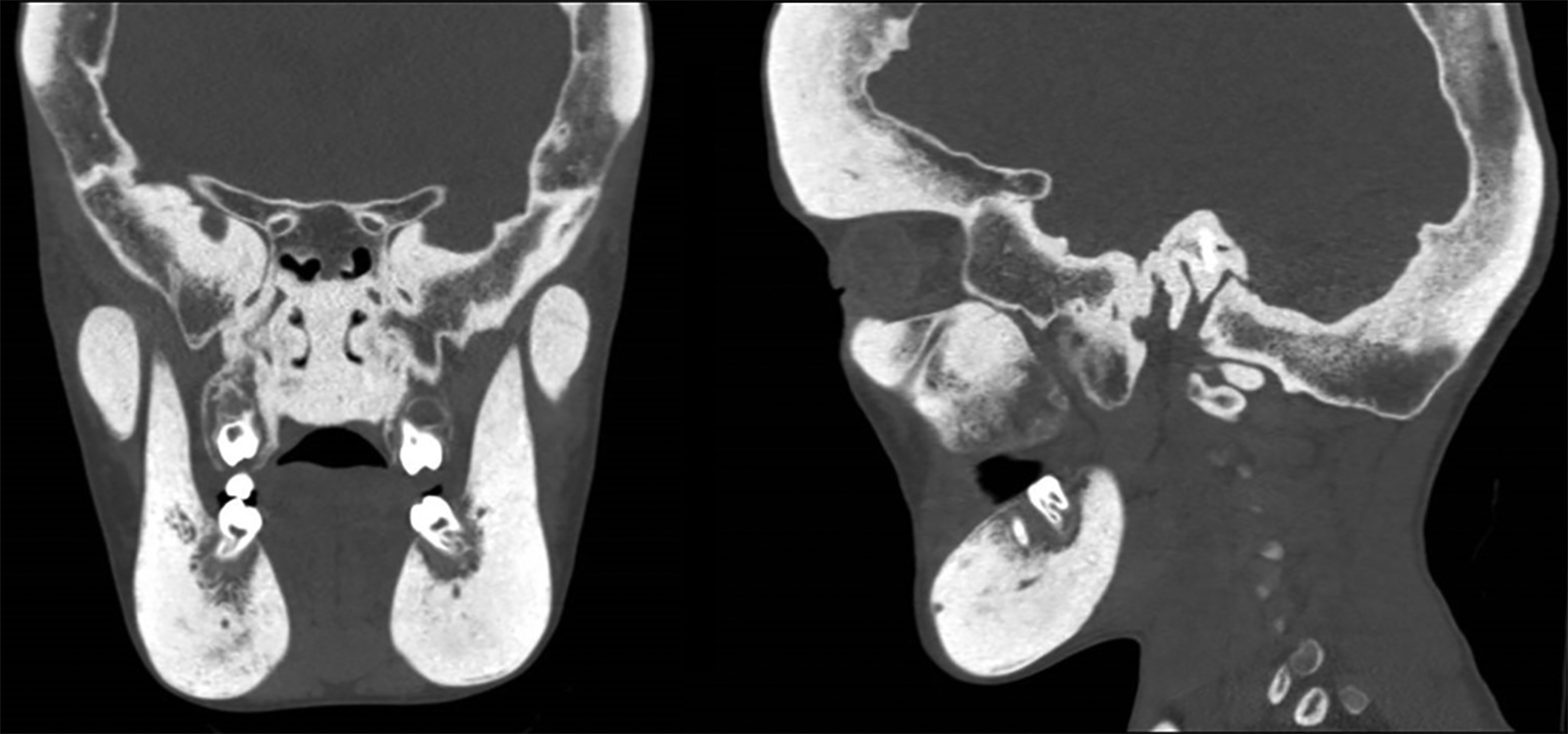
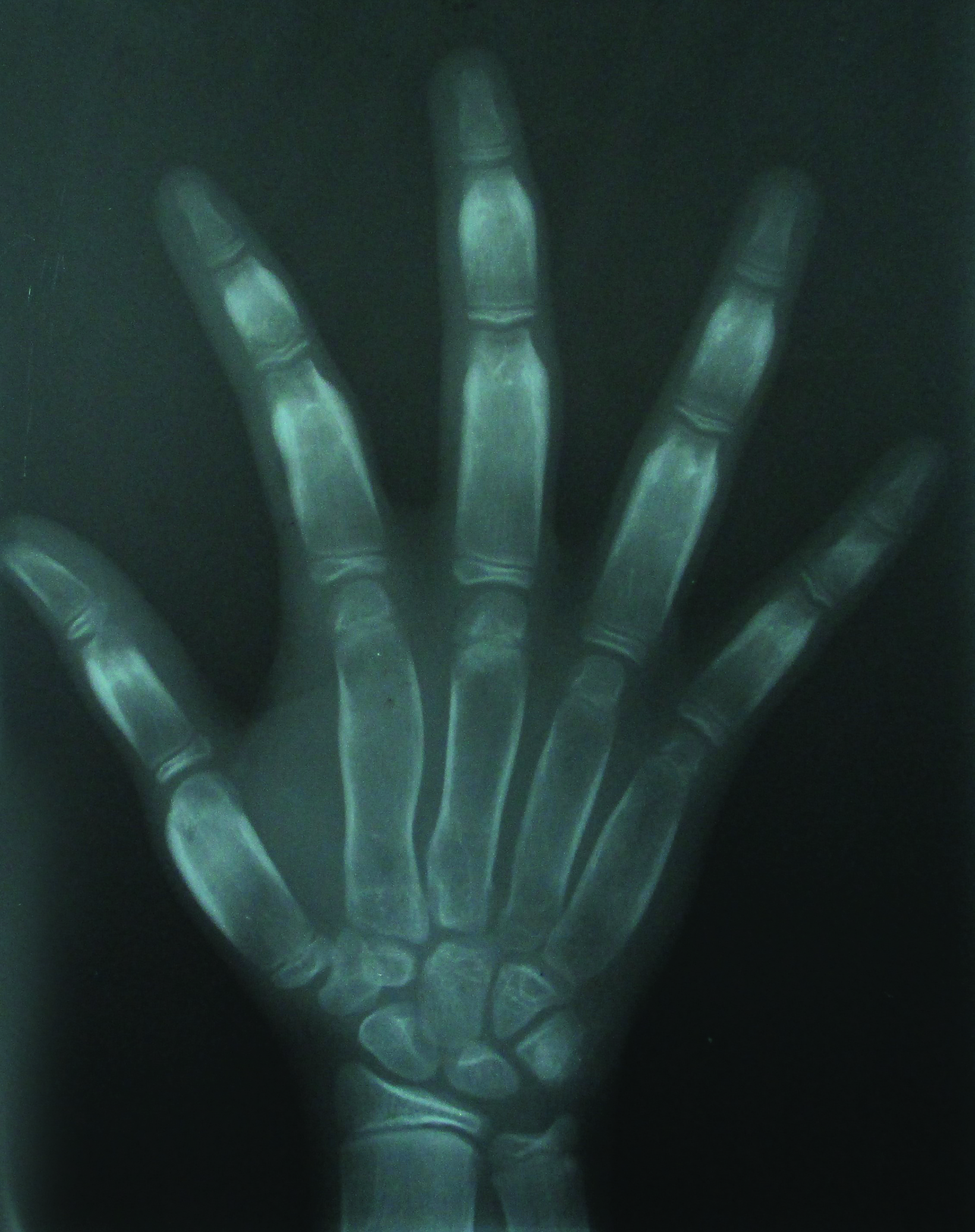
Axial, coronal, and sagittal computed tomography showed thickening of the calvarial and facial bones, widened diploic spaces, hyperostosis, and skull base sclerosis (Figures 2,3). A hand-wrist radiograph showed thickened cortices of all proximal phalanges (Figure 4).
Diagnosis
Craniometaphyseal dysplasia. Differential diagnosis includes osteopetrosis and craniodiaphyseal dysplasia.
Discussion
Craniotubular dysplasia consists of craniometaphyseal dysplasia (CMD) and Pyle disease.1
There are two types of CMDs: The autosomal dominant (AD, Jackson) and the autosomal recessive (AR). In the AD form, affected individuals generally have good health with unimpaired intellect. Progessive bone overgrowth may cause prognathism and malalignment of teeth. Sclerosis of the bone and overgrowth may lead to compression of the facial and vestibulococclear nerves.
Calvarial hyperostosis may result in increased intracranial pressure, and sclerosis may be manifested along the cranial sutures in adulthood. Obliteration of the sinuses, bossing of the frotonasal bone, hypertelorism, prognathic mandible, and widening of the
metaphyses of the long bones are the most common features of CMD. In the AR form, sclerosis and hyperostosis may be severe, with correspondingly severe cranial nerve compression, and the optic nerves may be involved. The AR form may also have pronounced facial palsy, loss of vision, and deafness.2
The widening of the metaphysis is more noticeable at the distal end of the femur. The flaring of the metaphyses results in an “Erlenmeyer flask” appearance in childhood, and a club-shaped deformity in adulthood.1
Mutation in ANKH gene, which is the human ankylosis gene, is thought to be the etiology of CMD. The mutation reduces the capacity to transport intracellular pyrophosphate from osteoblasts to the bone matrix.3 The AD form is linked to chromosome 5p15.2p14.1 and the AR form has been mapped to a 7cM interval on chromosome 6q21-22 (11, 14).1
Craniometaphyseal dysplasia should be differentiated from craniodiaphyseal dysplasia and osteopetrosis. Craniodiaphyseal dysplasia is characterized by massive, generalized hyperostosis and sclerosis, involving in particular the skull and facial bones. Athough most facial features are similar to CMD, they are more severe and pronounced. Unlike CMD, delayed milsestones, cardiac problems, and early death are common. Most patients die in the second decade of life. There is also evidence of significant sclerosis and flaring of the diaphyses and very little metaphyseal involvement.4
Osteopetrosis is a group of disorders in which the osteoclasts fail to resorb the primary spongiosa. This results in increased osseous density, which precludes imaging distinction between the cortical and cancellous bone. Histologically, there are an increased number of osteoclasts. This disorder has been subcategorized with a severe AR osteopetrosis called Albers-Schonberg disease, which can be considered as a differential diagnosis for CMD. Albers-Schonberg disease shows increased density of nearly all bones, along with associated complications including anemia, hepatomegaly, splenomegaly, blindness, deafness, facial paralysis, and osteomyelitis. Left untreated, patients with this disease have a high mortality rate. The epiphyses, metaphyses, and diaphyses are similarly affected.5
Patients with CMD require periodic follow-up examination for assessment of neurologic, ophthalmologic, and hearing difficulties, as bone thickening is progressive. Craniofacial surgery has a high failure rate. Surgical decompression may help to manage the rise in intracranial pressure. Medical management focuses on osteoclastic and osteoblastic activity modulation through calcitonin therapy or a low calcium diet supplemented by calcitriol.4
Conclusion
This case report highlights the pathognomonic clinical and radiological features of craniometaphyseal dysplasia and the differentiating factors of this entity from the other conditions categorized under osteochondrodysplasias. A multidisciplinary approach is essential to diagnosing and treating the condition to improve quality of life.
References
- Novelli G, Ardito E, Mazzoleni F, Bozzetti A, Sozzi D. An atypical case of craniometaphyseal dysplasia. Case report and surgical treatment. Ann Stomatol. 2017;VIII(2):89-94.
- Beighton P. Craniometaphyseal dysplasia ( CMD ), autosomal dominant form. J Med Genet. 1995;32:370-374.
- Thankappan S, Nair S. Craniometaphyseal dysplasia: A rare case in radiologic perspective. J Indian Acad Oral Med Radiol. 2014;26:428-431. doi:10.4103/0972-1363.155638
- Singh S, Qin C, Medarametla S, Hegde S V. Craniometaphyseal dysplasia in a 14-month old : a case report and review of imaging differential diagnosis. Radiol Case Rep. 2016;11(3):260-265. doi:10.1016/j.radcr.2016.04.006
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the Head and Neck, Fourth Edition. 2001.
References
Citation
M M, S B, SG MAB. Craniometaphyseal Dysplasia. Appl Radiol. 2022; (5):44-46.
August 31, 2022