Barth Syndrome: A Rare Cause of Congestive Heart Failure in Boys
Images
CASE SUMMARY
An otherwise healthy 14-month-old boy was admitted to the hospital with swelling in his feet and difficulty breathing related to upper respiratory infection symptoms. Work-up revealed congestive heart failure (CHF) with elevated serum brain natriuretic peptide (BNP) and troponin. Physical examination revealed full-appearing cheeks, small stature, and extremity hypotonia, as well as an inability to crawl or walk independently. He was found to be neutropenic with an absolute neutrophil count of 840. An echocardiogram was consistent with a dilated cardiomyopathy, with an ejection fraction of 30% and moderately dilated left ventricle.
This constellation of signs initiated further testing, including radiography; urine sampling, which revealed elevated 3-methylglutaconic acid, and a genetics evaluation. Genetic testing revealed a likely pathogenic variant identified in the TAZ gene. Serum testing showed elevated 3-methylglutaconic acid and pyruvate.
The patient was managed with treatment of CHF and hypertension. At 2 years of age the patient was noted to have short stature, (76.5 cm [<1% percentile]) and low body weight (8.7 kg [<1% percentile]). Of note, he was also found to have a preference for spicy foods, which has been described in this disorder.
IMAGING FINDINGS
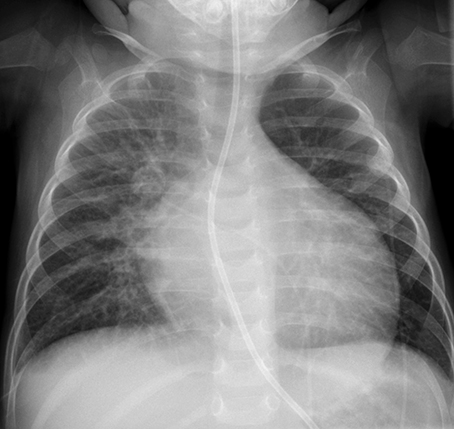
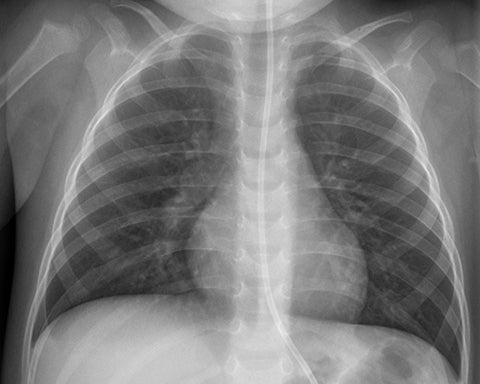
As noted above, echocardiography was consistent with a dilated cardiomyopathy. A portable chest radiograph (Figure 1) also demonstrated dilated cardiomyopathy, central vascular congestion, moderate interstitial edema with prominent Kerley B lines, consistent with congestive heart failure. Follow up chest radiograph (Figure 2) 9 months after therapy initiation showed resolution of cardiomyopathy and no residual findings of congestive heart failure.
DIAGNOSIS
Barth syndrome. Differential diagnoses include congenital heart disease, viral pneumonia, and Duchenne muscular dystrophy.
DISCUSSION
Barth syndrome (BTHS) is an X-linked recessive disorder caused by mutations in the TAZ gene (located on Xq28), which encodes for the enzyme tafazzin. Tafazzin functions to regulate remodeling of cardiolipin, which plays a role in cardiac function and mitochondrial ATP production; mutations in TAZ result in disorders in cardiolipin biosynthesis.1-3 This disorder in biosynthesis presents in tissues of the heart, liver, and skeletal muscle in particular, resulting in prolonged deficiency in ATP production, worsened by increased energy demand.2-5 Clinically, patients present with dilated cardiomyopathy, arrhythmias, neutropenia, and growth disturbance.4,6 Muscle weakness may present at birth, followed by delays in milestones such as crawling, hypotonia on examination, and difficulty with exercise and strenuous activity later in childhood.4 Because of the metabolic disturbance, patients may have a characteristic predilection for eating spicy foods.7 Males with the disorder are at increased risk for recurrent infections.4 There is no definite associated cognitive deficit.7
Fewer than 200 cases of BTHS have been described.6 Testing includes physical examination to assess growth milestones, muscle tone evaluation, and serum screening for markers of CHF. In addition, most patients present with neutropenia, which can be persistently low, intermittently normal to low, or cyclic.4 Echocardiography may show evidence of long-term left heart failure. Further testing with serum and/or urine studies will often reveal elevated lactic acid, creatinine kinase, and 3-methylglutaconic acid.6 Confirmation of diagnosis can be obtained by with targeted sequencing for mutations in the TAZ gene.
The long-term prognosis for Barth syndrome is variable. Infants have been reported to present with life-threatening sepsis as a result of neutropenia. Conversely, some males with the disorder may not present clinically until later in life. A multidisciplinary approach, including genetics counseling and involvement with cardiology, infectious disease, and gastroenterology specialists is required for management of symptoms.7
Though cases are sparse, patients are reported to have a reduced life expectancy and may succumb to heart failure in infancy. In some reported cases, the cardiomyopathy improves or resolves over time.4 Patients may also experience a catch-up growth phenomenon in adolescence.7 Overall reported life expectancy is into the late 40s.4
Treatments include medical management of neutropenia with granulocyte colony stimulating factor, dietary supplements, feeding aids, physical therapy, and psychiatric support.7 To this date there is no cure; potential therapies such as use of mitochondrial-targeted antioxidants have been explored.2
CONCLUSION
Barth syndrome is a rare cause of CHF in pediatric males. A constellation of findings including recurrent infections, neutropenia, CHF, and skeletal muscle hypotonia should warrant further genetics testing to evaluate for the disease. Mutations in the TAZ gene related to cardiolipin biosynthesis is the causative factor. While patients may be managed with medications for CHF, physical therapy, and factors to stimulate white blood cell growth, the disease course is guarded and life expectancy is affected.
REFERENCES
- Fatica EM, DeLeonibus GA, House A, Kodger JV, Pearce RW, Shah RR, Levi L, Sandlers Y. Barth syndrome: Exploring cardiac metabolism with induced pluripotent stem cell-derived cardiomyocytes. Metabolites. 2019; 9(12): 306.
- Saric A, Andreau K, Armand AS, Møller IM, Petit PX. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front Genet. 2016; Jan. 20; 6: 359.
- Dudek J, Maack C. Barth syndrome cardiomyopathy. Cardiovasc Res. 2017; 113(4): 399-410. doi: 10.1093/cvr/cvx014. Epub 2017 Feb 2
- Raja V, Reynolds CA, Greenberg ML. Barth syndrome: A life-threatening disorder caused by abnormal cardiolipin remodeling. J Rare Dis Res Treat. 2017; 2(2): 58-62. doi: 10.29245/2572-9411/2017/2.1087. Epub 2017 Mar 21.
- Ikon, N., Ryan, R.O. Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy. Lipids 52, 99–108 (2017). https://doi.org/10.1007/s11745-016-4229-7
- Finsterer J. Barth syndrome: mechanisms and management. Appl Clin Genet. 2019; 5(12): 95-106. doi: 10.2147/TACG.S171481. eCollection 2019.
- Reynolds S. Successful management of Barth syndrome: a systematic review highlighting the importance of a flexible and multidisciplinary approach. J Multidiscip Healthc. 2015; 8: 345–358.
Citation
L N, C B, AJ T, RB T,. Barth Syndrome: A Rare Cause of Congestive Heart Failure in Boys. Appl Radiol. 2021;(1):48-49.
January 19, 2021