Alobar Holoprosencephaly
Images
Case Summary
A routine obstetrical ultrasound detected multiple fetal brain and facial anomalies, as well as macrosomia, macrocephaly, and polyhydramnios, prompting further evaluation. Subsequent fetal MRI demonstrated severe brain and facial developmental abnormalities. A fetal echocardiogram was normal.
A liveborn infant was delivered via scheduled C-section. The neonate required intubation for respiratory failure and was found to have multiple endocrinopathies, including central diabetes insipidus, hypoglycemia, hypothyroidism, and growth hormone deficiency secondary to panhypopituitarism. The baby developed hypotension, persistent seizure activity, and worsening metabolic acidosis with episodes of prolonged bradycardia. Owing to declining clinical status, care was withdrawn.
Imaging Findings
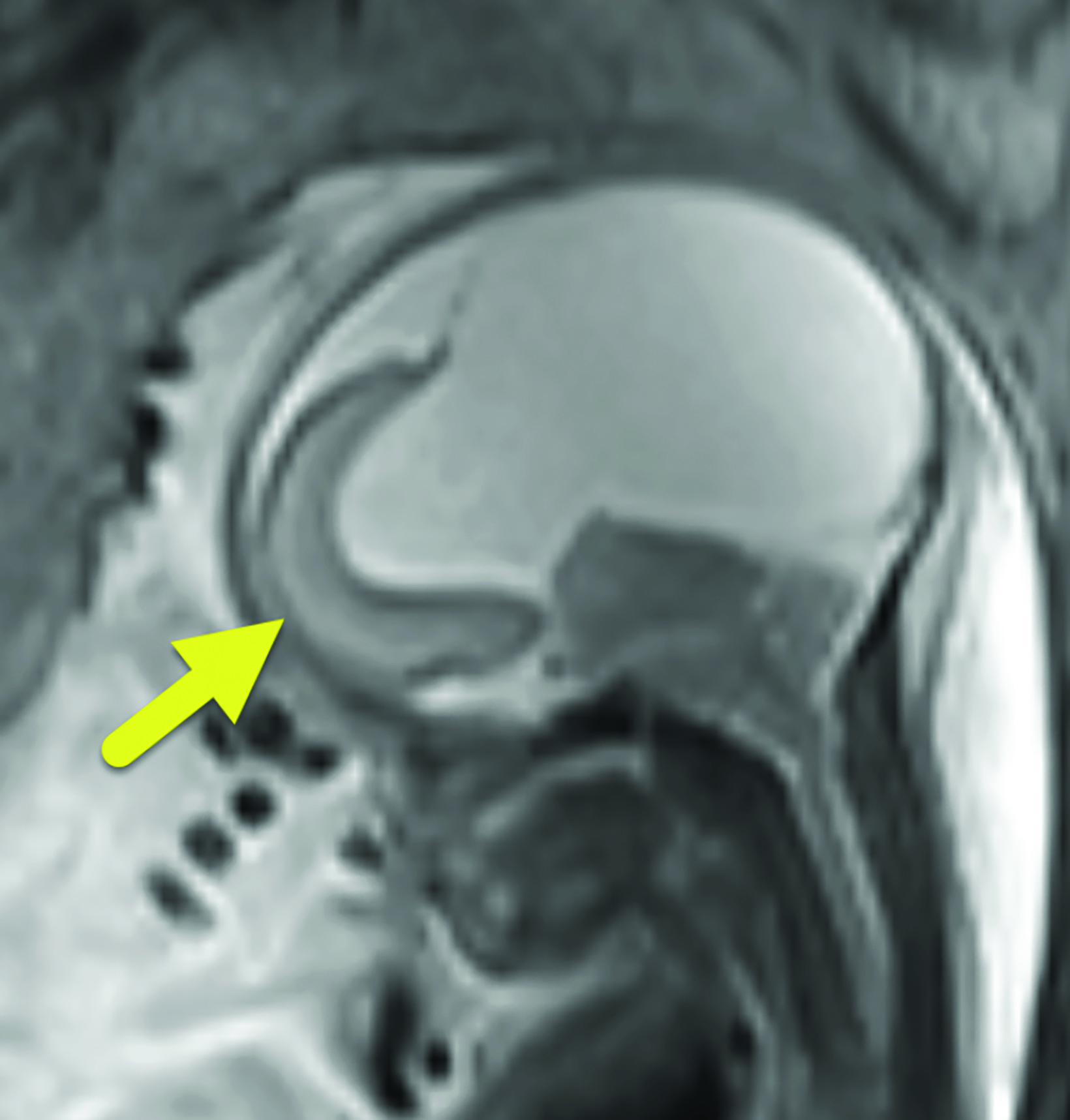
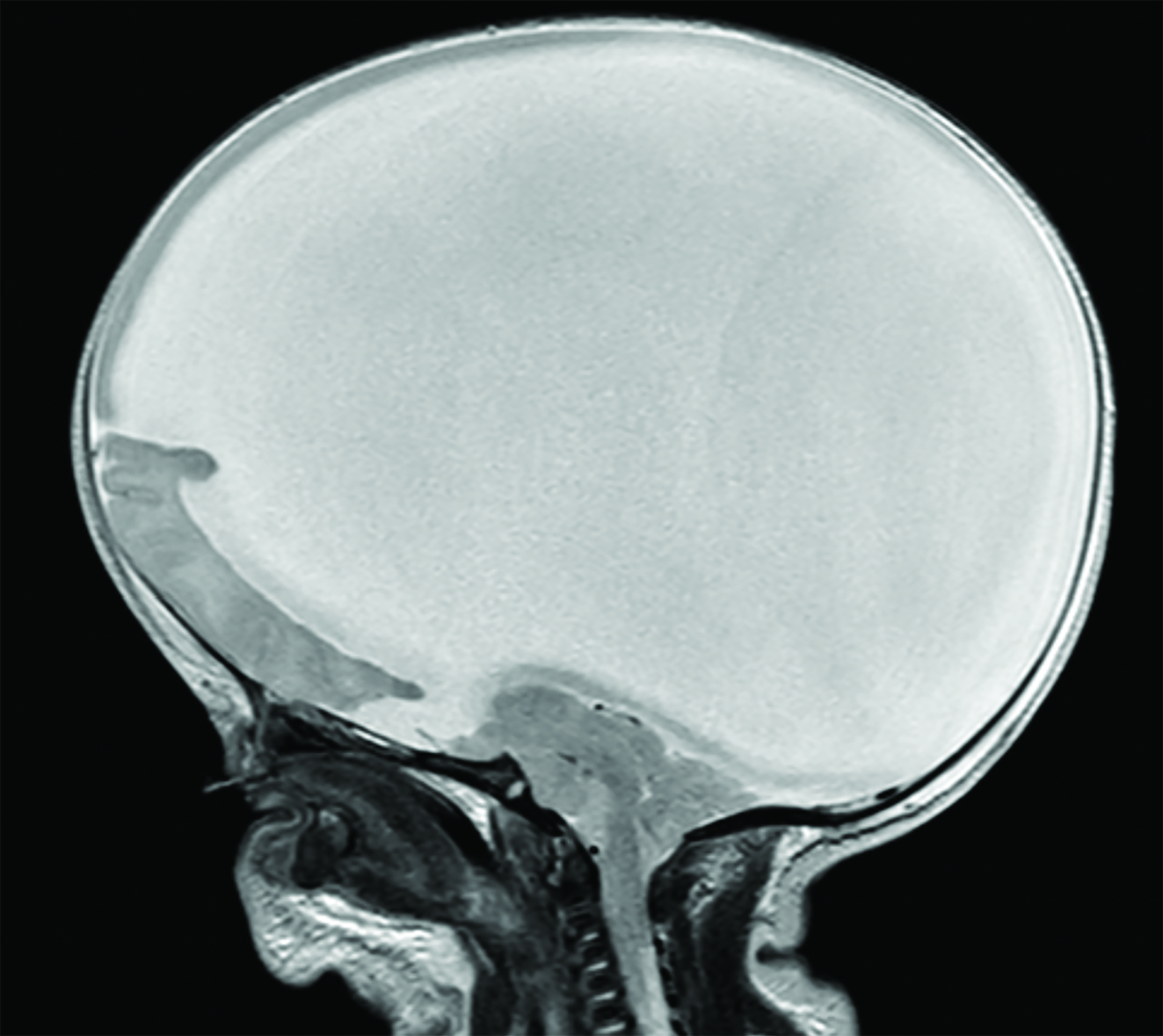
Fetal MRI (Figure 1) showed features of alobar holoprosencephaly (HPE), including failure of separation of the cerebral hemispheres and thalami, absence of normal supratentorial sulci, and a dilated supratentorial monoventricle communicating with a large dorsal cyst that caused mass effect on the posterior cranial fossa. The brainstem was severely dysplastic with incomplete segmentation between the thalami and the midbrain. There were midline cleft lip/palate and arrhinia with hypoplasia of the nasal cavity. Fetal eyes were not identified, suggesting severe bilateral microphthalmia or anophthalmia.
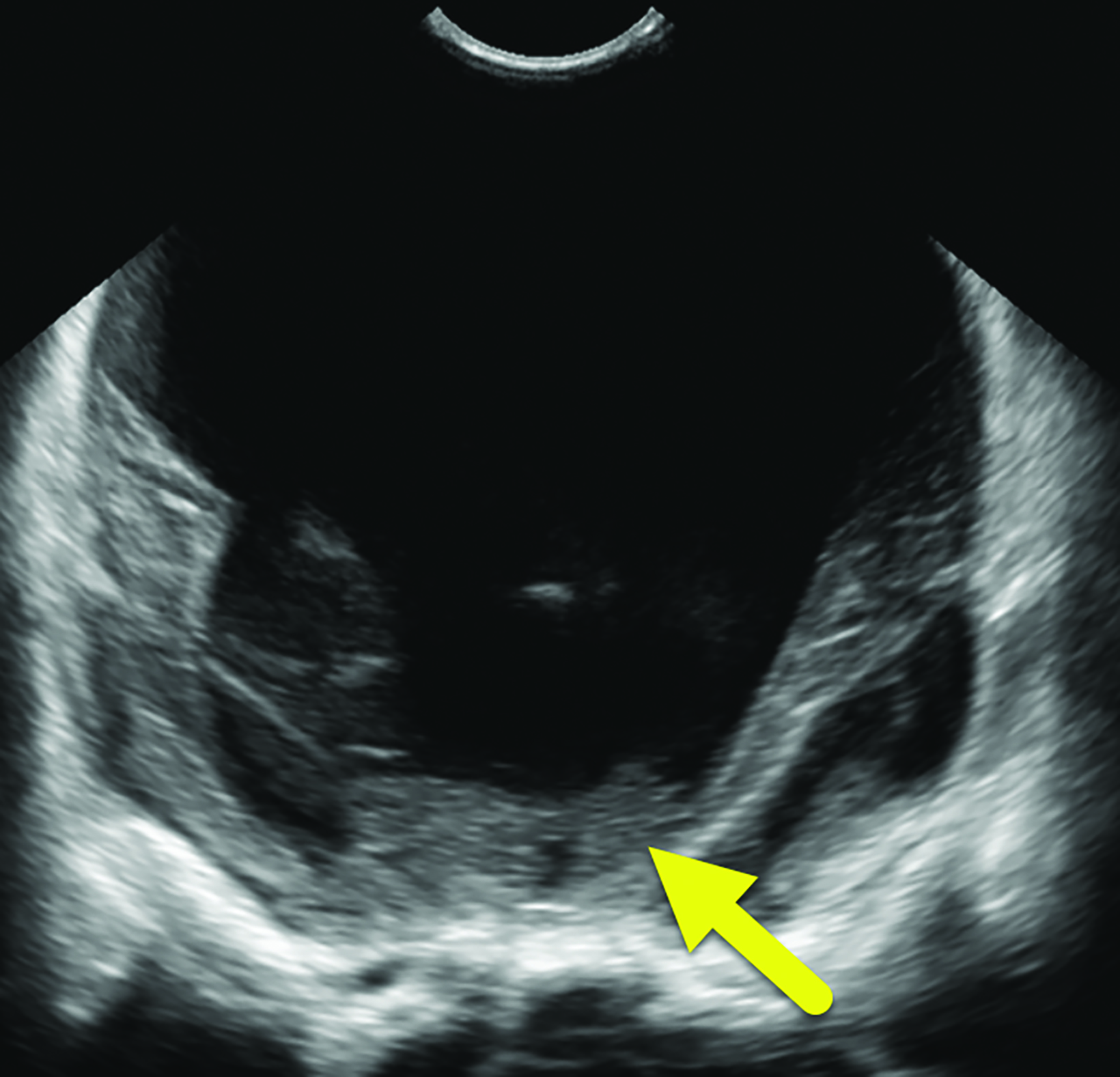
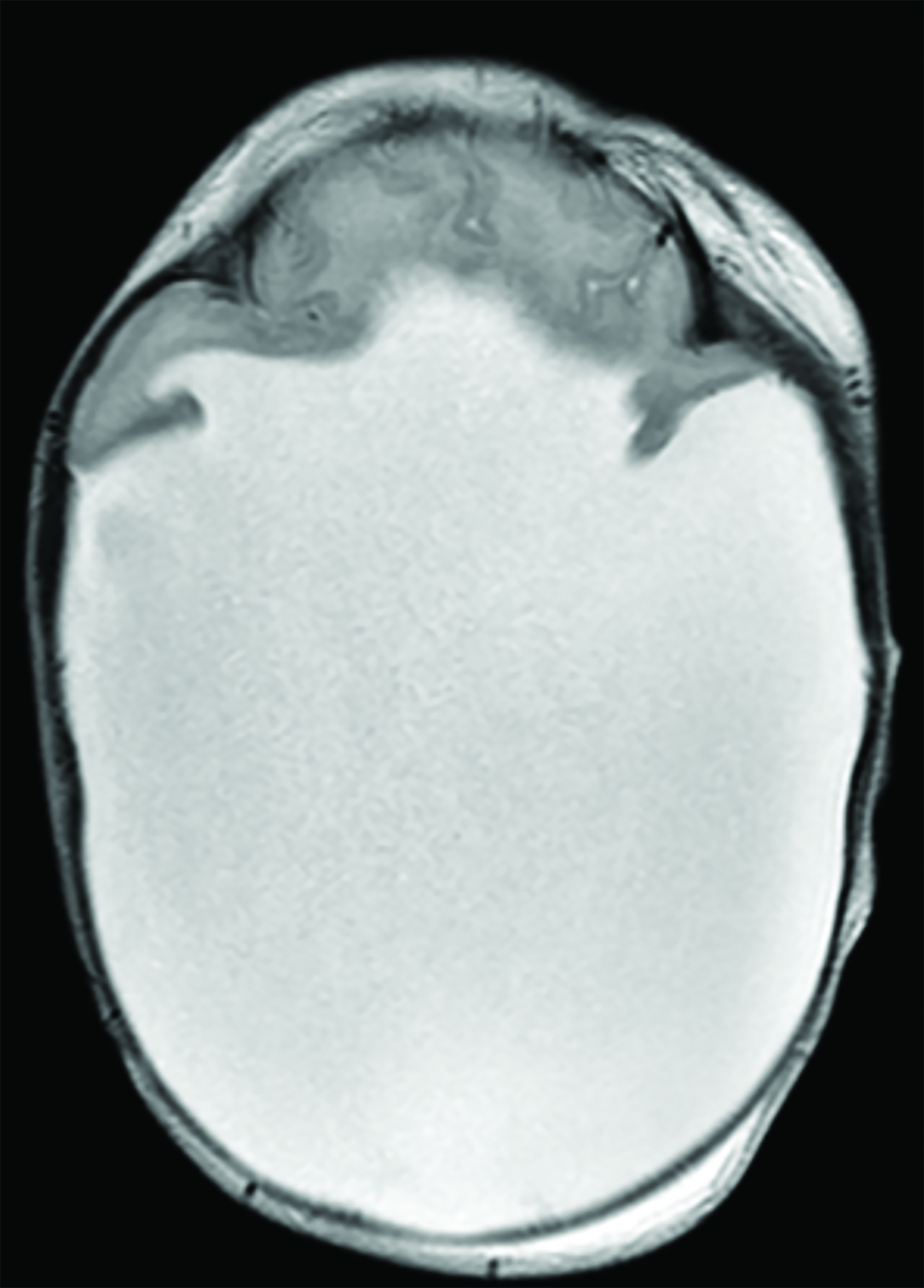
A postnatal head ultrasound (Figure 2) demonstrated a large monoventricle with absence of the corpus callosum, interhemispheric fissure, and cavum septum pellucidum.
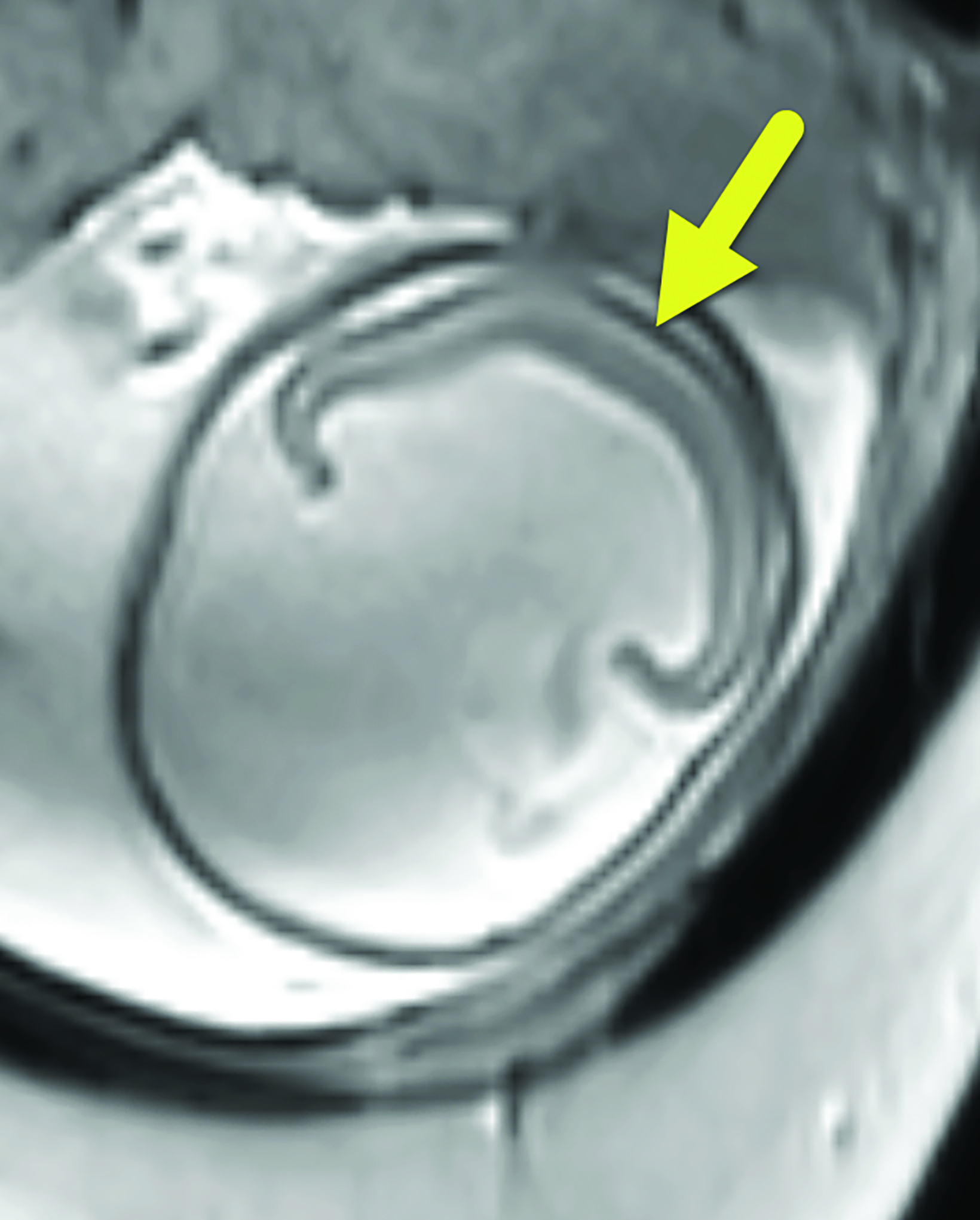
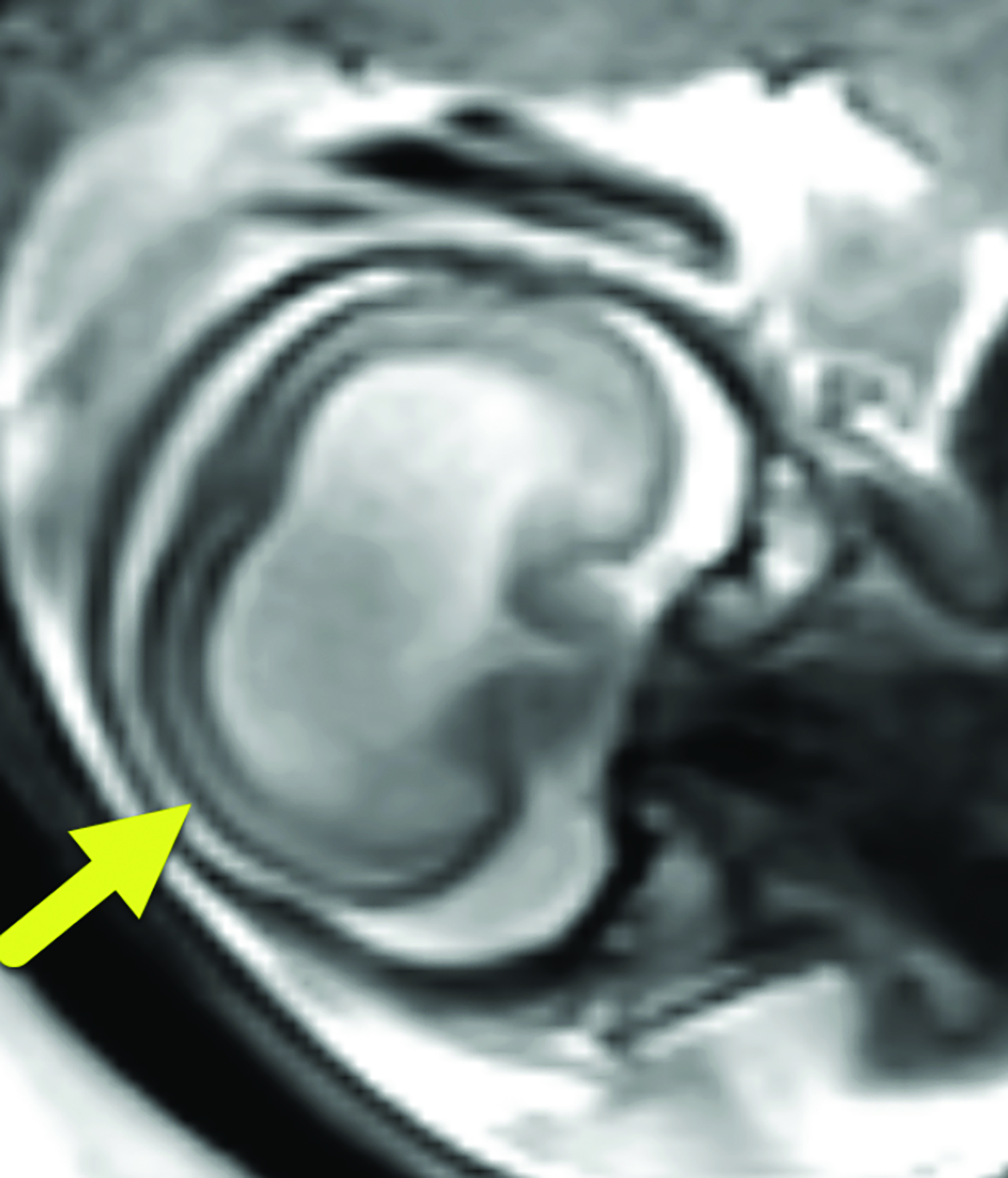
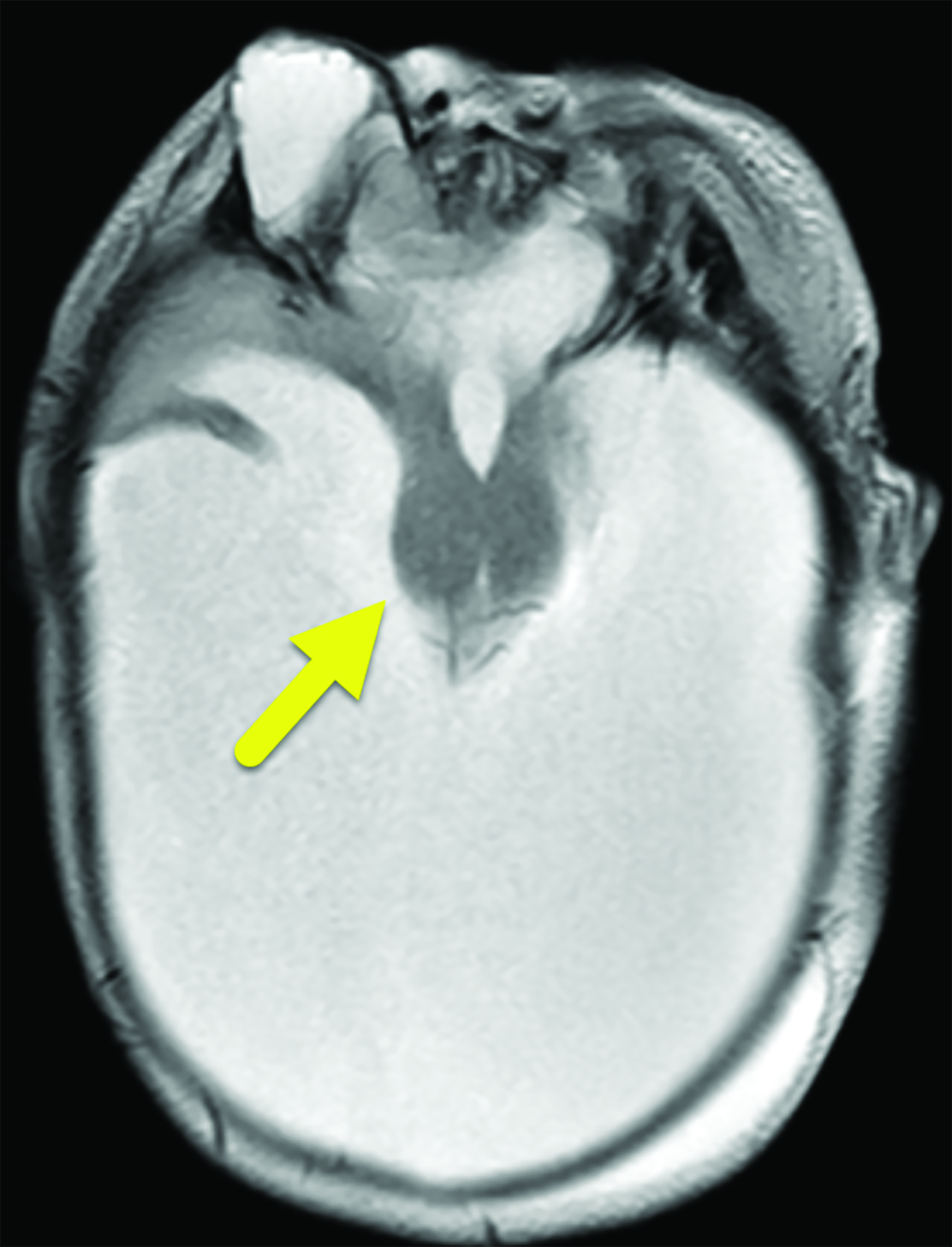
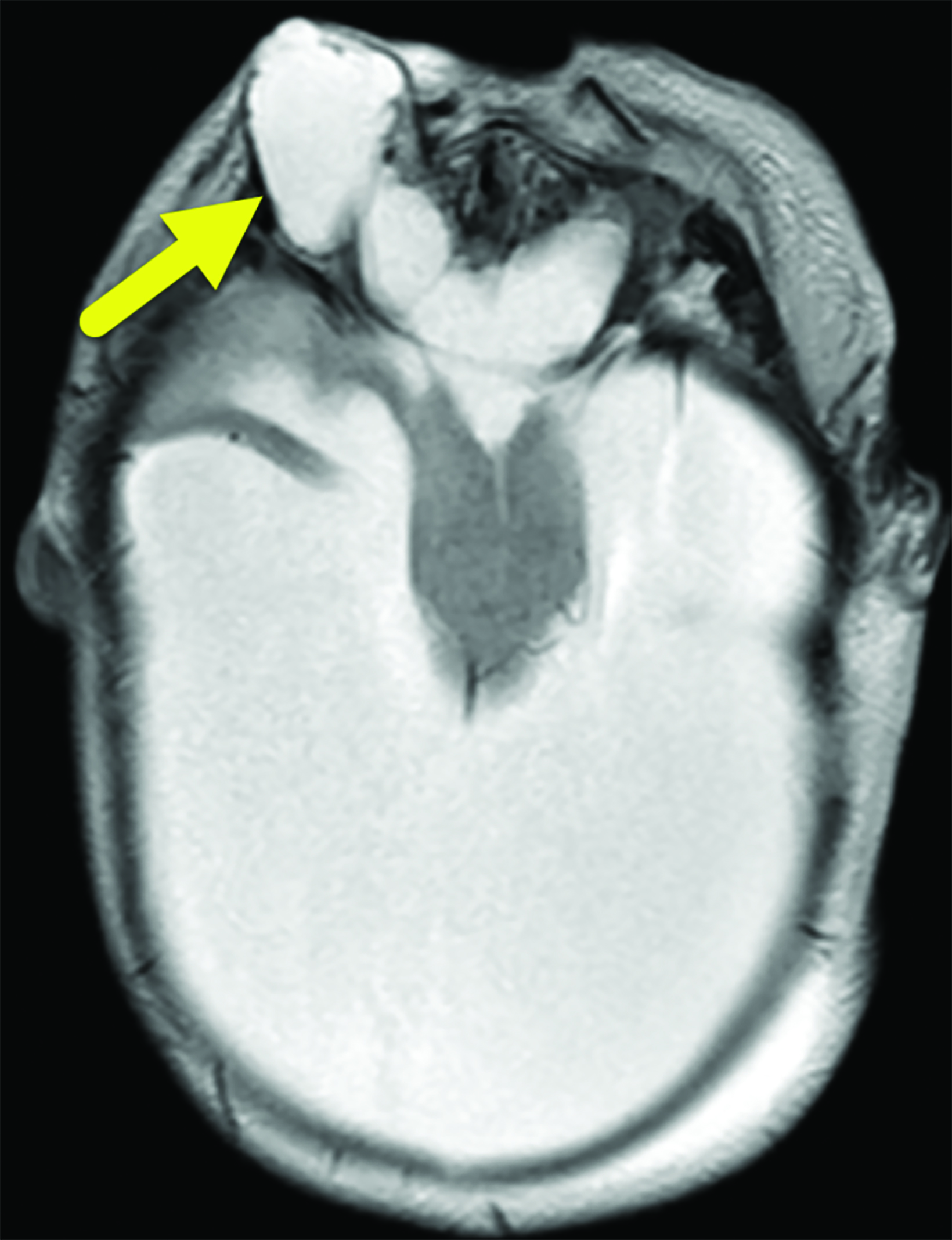
Brain MRI (Figure 3) showed intracranial findings of alobar HPE with fusion of frontal lobe parenchyma and the thalami, partial fusion of the midbrain, and absence of supratentorial midline structures. Additionally, there were features of macrocephaly and suspected aqueductal stenosis. The mass effect caused compression of posterior fossa structures with downward tonsillar hernia- tion into the cervical canal. The impaction of the cerebellar tonsils was worsened by marked stenosis at the craniocervical junction. Dysmorphic orbital and facial structures were noted, with cephaloceles extending into the orbits.
Diagnosis
Alobar Holoprosencephaly.
Differential diagnosis includes other subtypes of HPE: lobar, semilobar, and the middle interhemispheric variant.1
Discussion
Holoprosencephaly is a brain malformation that results from failed or incomplete division of the forebrain (prosencephalon) into right and left cerebral hemispheres between the 18th and 28th day of gestation.2 Incomplete separation of deep grey nuclei (thalami, hypothalamic nuclei, and basal ganglia) and malformation of cerebral ventricles is seen. HPE is estimated to occur in 1.31 of 10 000 live and stillbirths.3 Historically, HPE was classified into DeMyer’s three major subtypes based on severity: alobar, semilobar, and lobar HPE.4 A nonclassical subtype of HPE, known as the middle interhemispheric fusion variant, has since been characterized.5
Alobar HPE is the most severe form. Alobar HPE presents with complete or near complete nonseparation of the cerebral hemispheres and deep gray nuclei.4,5 In semilobar HPE, the anterior cerebral hemispheres fail to separate, but there is partial separation of the cerebral hemispheres posteriorly.4,5 Lobar HPE is the mildest subtype; the posterior cerebral hemispheres are separated and there is some separation of the anterior cerebral hemispheres.4,5 In the middle interhemispheric variant, the anterior frontal and occipital lobes are separated, but the posterior frontal and parietal lobes are not; the deep gray nuclei are typically separated.4,5
Prenatal ultrasound of the face and falx cerebri is the primary approach to diagnosing HPE as early as the first trimester.6, 7 Fetal MRI provides greater detail of developing brain structures.6, 7 Postnatally, head ultrasound is often the first modality utilized, since it can be performed at bedside. However, brain MRI is the gold standard for diagnosis and subtyping of the HPE spectrum. Serial imaging may be necessary in patients with enlarging head size.6
Alobar HPE has characteristic brain MRI findings such as fusion of the cerebral hemispheres, basal ganglia, hypothalamic nuclei, and thalamic nuclei.1, 5 There is a single midline, crescent-shaped monoventricle that often communicates with a dorsal cyst.1, 4, 5 The interhemispheric fissure, falx cerebri, corpus callosum, third ventricle and olfactory bulbs and tracts are absent.1
Newborns with alobar HPE commonly present with hypotonia, spasticity, and seizures.1 Hydrocephalus and macrocephaly may also be seen, owing to the presence of a large dorsal cyst and/or aqueductal stenosis; however, microcephaly is also common.1 Alobar HPE is often associated with a spectrum of severe midline craniofacial malformations. These include cyclopia with or without proboscis (tubular nose-like structure), ethmocephaly (separate orbits with proboscis), cebocephaly (closely spaced eyes with single-nostril nose), hypoplastic nose, hypotelorism, anophthalmia or microphthalmia, and bilateral cleft lip and palate.1, 5
Patients with alobar HPE are more likely to develop hydrocephalus and may benefit from a cerebrospinal fluid shunt.8 A gastrostomy tube is often placed to provide nutrition, as well as to reduce the risk of aspiration and recurrent lung infections.8,9
Alobar HPE is also associated with autonomic nervous system and hypothalamic dysfunction, which result in abnormal sleep-wake cycles, impaired body temperature regulation, and impaired thirst.8 These patients may develop life-threatening endocrinopathies. Central diabetes insipidus is the most common endocrinopathy in the setting of hypothalamic fusion and absent or dysfunctional hypothalamic-pituitary axis.10 Patients should also be evaluated for anterior pituitary hormone deficiencies such as hypothyroidism, hypoadrenocorticism, and growth hormone deficiency and given hormone replacement therapy as indicated.10 Prognosis of alobar HPE is very poor, with one study finding that one-half of patients die before 5 months, and 20–30% survive for at least 1 year.9
Conclusion
Alobar HPE is the most severe subtype of forebrain malformation. Characteristic brain MRI findings include fusion of cerebral hemispheres and deep gray nuclei in the rostral aspect of the skull, with a single midline ventricle that communicates with a dorsal cyst. Survival beyond infancy is poor, owing to the presence of severe neurologic impairment, seizures, craniofacial abnormalities, and multiple endocrinopathies.
Related Articles
References
Citation
A T, RB T, CM S, AJ T. Alobar Holoprosencephaly. Pediatric Supplement to Applied Radiology March/April 2024. 2024;(2):12-15.
March 5, 2024