A Primary Hepatic Neuroendocrine Carcinoma
Images
CASE SUMMARY
Primary neuroendocrine tumor of the liver is an exceedingly rare entity and is difficult to distinguish from other hepatic tumors based on radiographic findings. We report the case of a patient followed over a period of 3 years for a lesion in the left hepatic lobe. Upon hepatic lobe wedge resection, pathology revealed a primary hepatic neuroendocrine tumor.
IMAGING FINDINGS
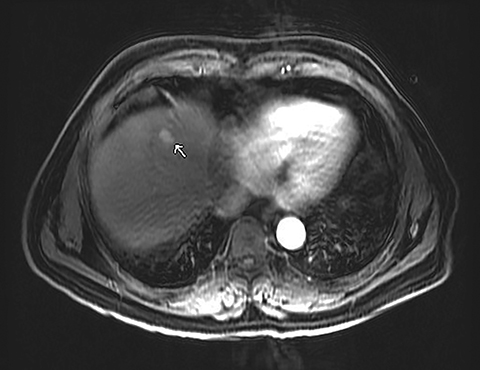
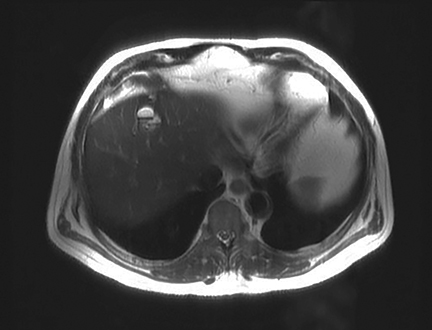
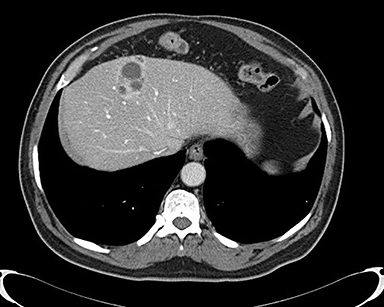
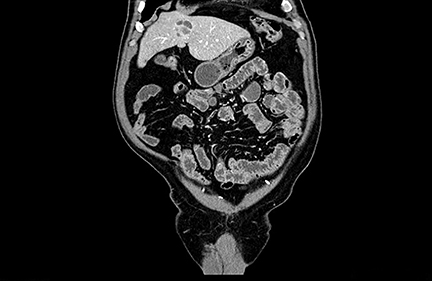
A 71-year-old patient presented with right upper-quadrant abdominal swelling. MRI of the abdomen demonstrated a 1.5 cm lesion in the left liver lobe (segment 4A) that was hyperintense on T2 imaging, demonstrated early arterial enhancement without washout (Figure 1), and showed diffusion restriction. A follow-up MRI at 6 months showed no significant change in the lesion. Subsequent follow-up MRI two years after presentation demonstrated an interval increase in lesion size to 2.5 cm in diameter. At this time, the lesion contained mixed solid and cystic components with fluid-fluid levels (Figure 2). Multiphase CT of the liver the following year showed continued interval growth of the mixed solid and cystic lesion. At that time, the lesion measured 4.4 cm in diameter (Figure 3).
The patient underwent evaluation for other primary malignancies. A CT of the chest, abdomen, and pelvis showed no evidence for other malignancy. Upper endoscopy was negative. Multiple polyps were discovered and several were biopsied during colonoscopy; none were malignant.
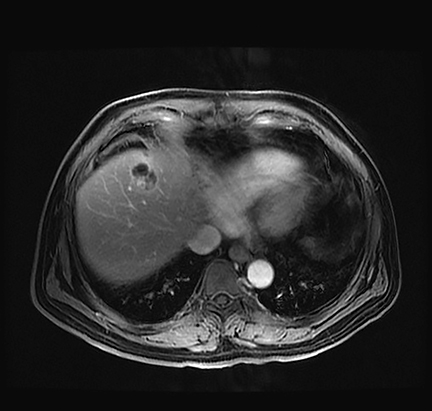
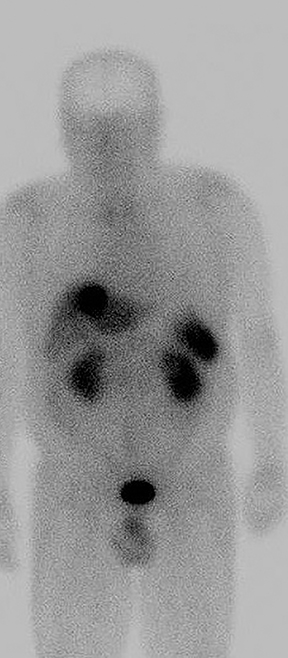
Suspicion for primary liver tumor or infection increased given the apparent absence of another primary malignancy. The patient was found to have an elevated serum chromogranin A level (204, subsequently 223; normal range: 25-140). Work-up for an infectious etiology, including Echinococcus Ab IgG EIA and Entamoeba histolytica IgG ELISA serologies, was negative. The elevated serum chromogranin A level raised suspicion for a neuroendocrine tumor. Subsequent nuclear medicine whole body octreotide In-111 scan demonstrated focal increased radiotracer uptake in the liver, corresponding to the lesion seen on CT and MRI (Figure 4).
The patient underwent liver wedge resection. Immunohistochemical evaluation demonstrated positivity for chromogranin and synaptophysin markers, considered specific markers for neuroendocrine tumors.
DIAGNOSIS
Primary hepatic neuroendocrine carcinoma
DISCUSSION
Neuroendocrine tumors, which arise from neuroendocrine cells, are rare. They can arise in any organ that contains peptide and amine producing cells, but most occur in the gastrointestinal tract and lungs.1 Diagnosis of neuroendocrine tumors is confirmed by pathology. Specific immunohistochemical markers include chromogranin A and synaptophysin. Octreotide imaging has a sensitivity up to 90% for neuroendocrine tumors.2 Most of these tumors involving the liver are metastases from another primary source. They tend to demonstrate strong arterial enhancement due to their vascularity.3
Primary hepatic neuroendocrine carcinoma (PHNEC) is an exceedingly rare entity, with only a few hundred cases reported in the literature.4,5 PHNEC shows no strong gender predominance, but it tends to occur in patients aged 40-50 years.6 PHNEC is often asymptomatic, or it may present with nonspecific symptoms such as abdominal pain, bloating, or jaundice.
Owing to the small number of reported cases, the imaging characteristics of PHNEC are not well defined and are difficult to distinguish from other primary tumors of the liver and metastases. Many reports have demonstrated cystic components on MRI and CT imaging,5 similar to our case. In a case of suspected PHNEC, extrahepatic neuroendocrine tumors should be excluded with upper and lower endoscopy, as well as with CT and PET imaging.
Treatment of PHNEC depends on the location, aggressiveness, and presence of metastases. No staging guidelines currently exist for PHNEC. If it is confined to the liver, surgical resection must be considered. If the tumor is unresectable, transarterial chemoembolization is a potential treatment option. If metastases are present, chemotherapy is another potential management option; however, outcomes vary.4
There are currently no characteristic features to reliably differentiate PHNEC from other liver tumors. If PHNEC is suspected, serum markers, including chromogranin A levels, should be checked. Additionally, octreotide imaging can be obtained to assess the neuroendocrine nature of the tumor. A thorough investigation for an extrahepatic primary neuroendocrine tumor is necessary to exclude an etiology for metastasis, which is much more frequently encountered.
REFERENCES
- Yao JC, Hassan M, Phan A et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008; 26: 3063-3072.
- Crithchley M. Octreotide scanning for carcinoid tumors. Postgrad Med. J. 1997; 73: 299-402.
- Horton KM, Kamel I, Hofmann L, Fishman EK. Carcinoid tumors of the small bowel: a multitechnique imaging approach. AJR Am J Rentgenol. 2004; 182 (3): 559-567.
- Park CH, Chung JW, Jang SJ, Chung MJ, Bang S, Park SW, Song SY, Chung JB, Park JY. Clinical features and outcomes of primary hepatic neuroendocrine carcinomas. J Gastroenterol Hepatol. 2012 Aug; 27(8):1306-1311.
- Zi-Ming Zhao, Jin Wan, Ugochukwu C. Ugwuowo, Liming Wang, Jeffrey P. Townsend. Primary hepatic neuroendocrine carcinoma: report of two cases and literature review. BMC Clin Pathol. 2018;18:3.
- Quartey B. Primary hepatic neuroendocrine tumor: what do we know now? World J Oncol. 2011; 2(5): 209-216.
Citation
DM B, P K,. A Primary Hepatic Neuroendocrine Carcinoma. Appl Radiol. 2021;(2):38-40.
March 11, 2021