Wolman disease
Images





CASE SUMMARY
A 1-month-old female presented to our institution with a history of poor feeding, diarrhea and a 2-lb weight loss. Upon evaluation, the patient was determined to have severe dehydration, acute renal failure, and metabolic acidosis. Cornelia De Lange syndrome was initially suspected by the geneticists due to dysmorphic facial features, including prominent eyebrows, thin upper lip, and smooth philtrum. Imaging revealed bilateral adrenal enlargement and calcifications with maintenance of the adreniform shape. Bone marrow findings and genetic tests were performed and confirmed the diagnosis.
IMAGING FINDINGS
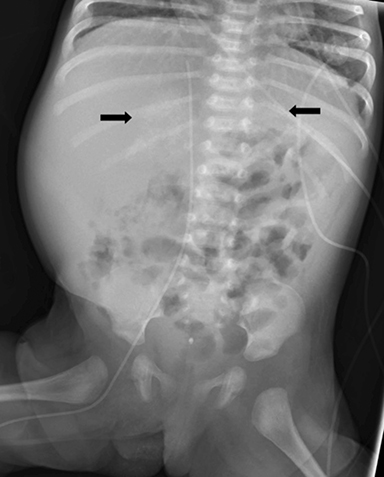
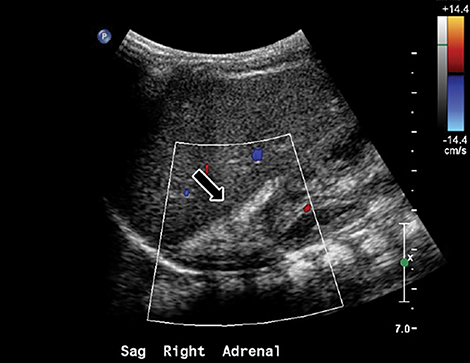
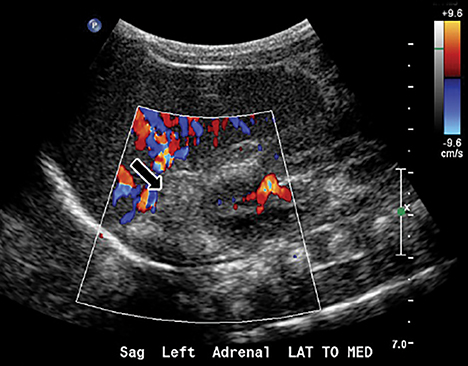
Initial plain radiograph of the abdomen for catheter placement demonstrated dense calcifications in the region of the adrenal glands (Figure 1). Subsequent abdominal ultrasound showed bilateral enlarged and calcified adrenal glands (Figures 2 and 3) which maintained their adreniform shape. A CT scan of the abdomen and pelvis with and without contrast showed bilaterally enlarged calcified adrenal glands, hepatomegaly with fatty change, splenomegaly, and mild ascites (Figures 4-6).
DIAGNOSIS
Wolman disease
The differential diagnoses include neuroblastoma, ganglioneuroblastoma, adrenal hemorrhage, granulomatous disease/infection, lymphoma, and adrenal carcinoma.
DISCUSSION
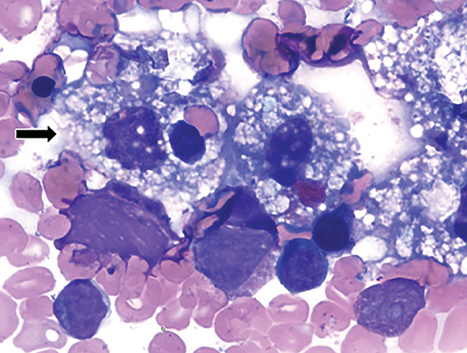
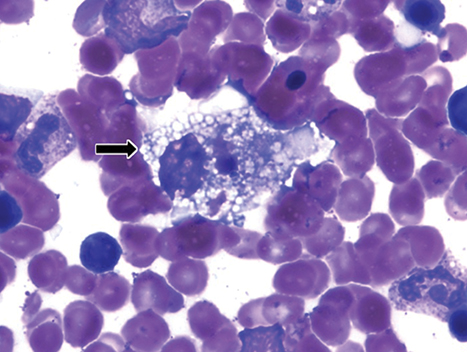
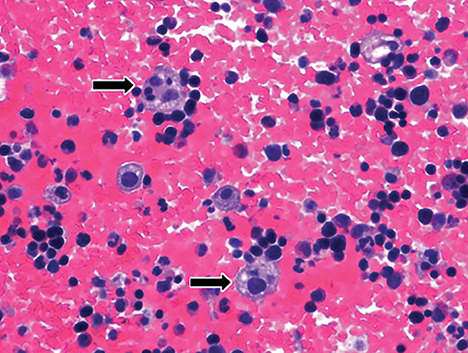
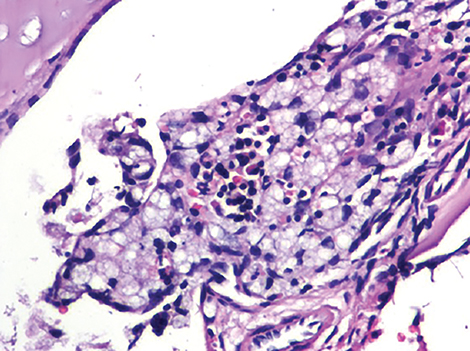
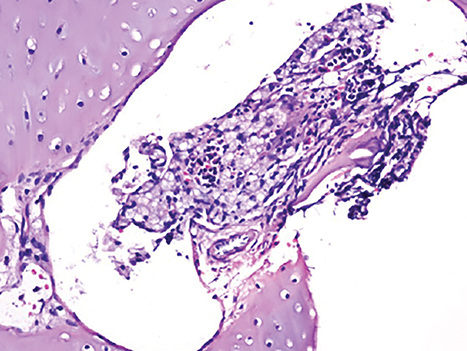
Wolman disease was first described by Moshe Wolman, a neuropathologist, in 1956.1 It is a rare autosomal recessive inborn error of lipid metabolism (the gene for this enzyme is located on chromosome 10q23.2-q23.3.2) characterized by lysosomal acid lipase deficiency. This inborn error of metabolism leads to massive accumulation of cholesteryl esters and triglycerides, particularly within the reticuloendothelial system, the mucosa and outer layers of the small intestine, and the adrenal cortex.2,3 Cells of the reticuloendothelial system accumulate lipids and transform into foam cells (Figures 7-13), which leads to hepatosplenomegaly and lymphadenopathy. Bone marrow may show foamy macrophages (Figures 7-13). Definitive diagnosis is made by biochemical demonstration of deficient lysosomal acid lipase activity.4 Onset in older patients has been named cholesteryl ester storage disease.4
Clinical symptoms are nonspecific and can include vomiting, steatorrhea, failure to thrive, poor weight gain, abdominal distension, and abnormal neurological development. Hepatosplenomegaly can be seen due to lipid accumulation within these organs, although lipid accumulation is widespread. Interestingly, it is usually the adrenal glands that are solely calcified, with sparing of the adrenal medulla.
Bilateral adrenal enlargement and calcification are seen and confined to the zona reticularis and inner fasciculata, with sparing of the zona glomerulosa, outer fasiculata and medulla.5 This pattern is pathognomonic for Wolman disease. Cortical location of calcifications is likely due to high uptake of low-density lipoproteins by the adrenal steroid synthesizing cells with subsequent necrosis. This may prevent the adrenal glands from producing essential hormones that can affect blood pressure, metabolism and the immune system. The small intestine is usually dilated with thickened walls, characterized by infiltration of foamy histiocytes into the lamina propria of the small intestine and even the colon.6
Plain radiographs can demonstrate bilateral adrenal calcifications as well as hepatomegaly. Renal ultrasound can demonstrate dense calcifications in the enlarged adrenal glands, with maintenance of the adreniform shape. CT can show dense adrenal calcifications and adrenal enlargement. Additionally, hepatosplenomegaly with hepatic steatosis can be demonstrated due to lipid accumulation. There is usually no displacement or distortion of the adjacent structures. MRI may show hepatosplenomegaly and bilateral adrenal gland enlargement. Bilateral adrenal calcifications will show low signal foci on both T1- and T2-weighted images.6,7
The differential diagnosis for adrenal calcification is extensive. A distinguishing feature that favors the diagnosis of Wolman disease includes uniform bilateral enlargement and cortical calcifications. Other conditions that cause adrenal calcifications result in: a) decreased or normal size (adrenal hemorrhage or granulomatous disease), b) variation of shape and size of the adrenal gland, c) or adjacent renal displacement (tumors).
Wolman disease is almost always fatal in the first year of life, with malabsorption and diarrhea representing the main cause of demise. Widespread lipid accumulation can lead to multi-organ failure. Treatment is focused on reducing specific complications. A fatty-ester-free diet may help with steatorrhea. Bone marrow transplantation has had mixed results.
CONCLUSION
Wolman disease is a rare condition that affects neonates due to an inborn error of lipid metabolism. Radiological findings of bilateral adrenal enlargement and cortical calcifications with maintenance of the adreniform shape are pathognomonic. It is important to recognize the disease because it is often fatal in the first year of life.
REFERENCES
- Wolman M, Sterk V, Gatt S et al. Primary familial xanthomatosis with involvement and calcification of adrenals: report of two more cases in siblings of a previously described infant. Pediatrics. 1961; 28:742-757.
- Anderson R, Byrum R, Coates P et al. Mutations at the lysosomal acid lipase/cholesteryl esterase (LIPA) deficient in wolman disease to chromosome 10q23.2-q23.3. Genomics.1993; 15: 245–247.
- Nchimi A, Rausin L, Khamis J. Ultrasound appearance of bowel wall in wolman’s disease. Pediatric Radiology.2003; 33:284-285.
- Reiner Z, Guardamagna O, Nair D et al Lysosomal acid lipase deficiency – an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014; 235: 21-30.
- Dutton R. Wolman’s disease ultrasound and CT diagnosis. Pediatric Radiology. 1985;15:144-146.
- Low G, Irwin G, MacPhee G et al. Characteristic imaging findings in wolman’s disease. Clin Radiol Extra. 2004; 59:106-108.
- Ozmen M, Aygun, N, Kilic I et al Wolman’s disease: Ultrasonographic and computed tomographic findings. Pediatric Radiology. 1992;22: 541-554.