Primary Langerhans cell histiocytosis of the temporal lobe
Images

Primary Langerhans cell histiocytosis of the temporal lobe
Findings
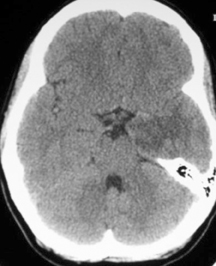
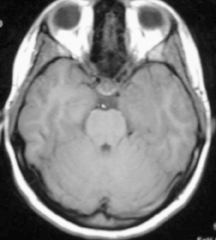
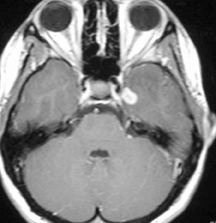
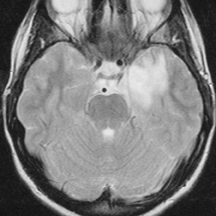
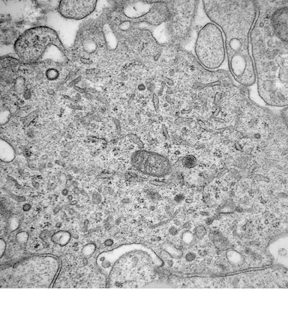
A non-contrast computed tomography (CT) scan demonstrated an ill-defined area of decreased attenuation within the left temporal lobe (Figure 1). Post-contrast images revealed this to be a 1 × 1.1 cm rim-enhancing lesion (Figure 1). Subsequent magnetic resonance (MR) imaging showed a 13-mm left temporal lesion that was hypointense and ring-enhancing on T1-weighted imaging (Figures 2). The mass was hyperintense on T2 FLAIR with extensive vasogenic edema and local mass-effect (Figure 2). CT scans of the chest, abdomen, and pelvis were negative, and a whole-body bone scan showed no osseus lesions. Microscopic examination of the resected specimen demonstrated sheets of large cells with lobulated nuclei and an immunohistochemistry profile reactive for CD1a and S100. Electron microscopy was significant for the presence of numerous Birbeck granules (Figure 3). These combined features were diagnostic of Langerhans cell histiocytosis.
Discussion
Langerhans cell histiocytosis (LCH) is a group of rare and diverse disorders defined by the pathologic infiltration and proliferation of Langerhans cells. Previously known as histiocytosis X, the disorder’s name was changed to LCH as the primary cell involved was elucidated.
LCH encompasses a spectrum of clinical manifestations ranging from a solitary chronic lesion, known as an eosinophilic granuloma, to a fulminant multisystem process called Letterer-Siwe disease. An intermediate form, Hand-Shüller-Christian disease, is composed of the triad of calvarial lesions, diabetes insipidus, and exophthalmos.
The pathophysiology of LCH is incompletely understood, with disagreement over whether the mechanism is a reactive or neoplastic one.1 The occurrence of spontaneous remissions and the absence of karyotypic abnormalities support a reactive progression. On the other hand, studies demonstrating monoclonality and response to chemotherapeutic agents lend support to a neoplastic process.
Although the brain is often secondarily involved in systemic disease, solitary lesions of the brain parenchyma, outside the hypothalamic–pituitary axis, are exceedingly rare. Only 14 cases have been reported in the literature. Including the current patient, the median age of affected individuals is 23.2 ± 11.2 years. Males predominate, with the current case being only the fourth female patient. Most lesions involve the temporal lobe, although some also involve the parietal and frontal lobes. The most common presenting complaint is seizure, with every patient reporting at least one episode. Other common symptoms include headache and hemiparesis.
CT findings generally reveal a low-density mass, although 2 cases reported isodense2 and hyperdense3 lesions. The lesions are contrast-enhancing with either a ring or a homogenous pattern.
Only 6 papers have described MR findings. T1-weighted images typically demonstrate low-intensity or isointense lesions while T2-weighted images show high-intensity lesions. In all cases except one, lesions enhanced with gadolinium.2
Definitive diagnosis always requires histological evaluation. The diagnostic Langerhans cell (LC) is a large mononuclear cell with a folded nucleus resembling a coffee bean. The Birbeck granule, a trilaminar rod-like organelle with a terminal expansion resembling a tennis racquet, is distinctive. Immunohistochemical staining can help confirm the diagnosis with reactivity toward the S100 and CD1a antigens.
The pathophysiology of central nervous system (CNS) LCH has not been well studied. Although clinical manifestations can be partially attributed to mass effect, neurodegeneration characterized by neuronal and axonal destruction with secondary demyelination4 has also been described.
While multisystem LCH responds to various chemotherapeutic regimens,5 the treatment of intracranial LCH is not yet well established. A retrospective study looking at histologically documented CNS LCH reported that localized lesions could be treated successfully with either surgery or radiation, while multiple lesions could be controlled with chemotherapy.6 Notably, gamma knife radiosurgery has been successful in excising a pontine lesion.7 Long-term prognosis following resection has been good, with lesions recurring in only 2 patients.7,8 No cases progressed to systemic disease.
Conclusion
Solitary LCH involving the cerebral hemispheres is a rare entity with only 14 cases reported in the literature. CT findings are generally of a contrast-enhancing, low-density mass. On MR, T1-weighted images typically demonstrate a low-intensity or isointense lesion while T2-weighted images show a high intensity lesion that enhances with gadolinium. The authors recommend that LCH be considered in the differential diagnosis when a patient presents with new-onset recurrent seizures and suggestive radiographic findings.
- Gonzalez CL, Jaffe ES. The histiocytoses: Clinical presentation and differential diagnosis. Oncology (Williston Park). 1990;4:47-60; discussion 60, 62.
- Penar PL, Kim JH, Chyatte D. Solitary eosinophilic granuloma of the frontal lobe. Neurosurgery. 1987;21: 566-568.
- Itoh H, Waga S, Kojima T, et al. Solitary eosinophilic granuloma in the frontal lobe: Case report. Neurosurgery. 1992;30:295-298.
- Grois N, Prayer D, Prosch H, et al. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain. 2005;128(Pt 4):829-838.
- Egeler RM, de Kraker J, Voute PA. Cytosine-arabinoside, vincristine, and prednisolone in the treatment of children with disseminated Langerhans cell histiocytosis with organ dysfunction: Experience at a single institution. Med Pediatr Oncol. 1993;21:265-270.
- Hund E, Steiner H, Jansen O, et al. Treatment of cerebral Langerhans cell histiocytosis. J Neurol Sci. 1999;171:145-152.
- Cagli S, Oktar N, Demirtas E. Langerhans’ cell histiocytosis of the temporal lobe and pons. Br J Neurosurg. 2004;18:174-180.
- Sivalingam S, Corkill G, Ellis WG. Focal eosinophilic granuloma of the temporal lobe. Case report. J Neurosurg. 1977;47:941-945.
Related Articles
Citation
Primary Langerhans cell histiocytosis of the temporal lobe. Appl Radiol.
February 8, 2012