Pediatric neuroradiology, part 2: Embryologic basis for inherited neurological disease and congenital neoplasm
Images




Knowledge of basic brain embryology provides the foundation for making diagnoses of brain malformations, heritable diseases, congenital neoplasms and even disorders of postnatal development.
In the July/August issue (38;7-8:29-40), I discussed how an understanding of basic brain embryology provides the basis for a more thorough understanding of these pathologic processes.
In this article, I discuss heritable neurological diseases that are caused by genetic errors leading to defects in the normal processes of brain formation. These defects typically have imaging stigmata that, when learned, are easily recognizable. Congenital brain neoplasms, malformations and other neurological diseases may be associated with hydrocephalus and can develop at almost any time of brain development. Recognition of the imaging milestones in postnatal brain maturation, primarily the process of myelination, is important in differentiating dysmyelination from degenerative processes.
Innumerable neurological diseases are inherited through dominant and recessive genetic traits. In this article, the most commonly encountered diseases and syndromes, with radiographic manifestations, are reviewed.
Phakomatoses
The phakomatoses are a group of congenital malformations of the ectodermal tissues. The neuroectodermal manifestations are generally due to anomalies of neuronal proliferation, differentiation and histiogenesis.1 These syndromes are the result of inherited chromosomal disorders, are sometimes due to spontaneous chromosome mutations or may be degenerative. Although there are numerous syndromes and diseases that fit the diagnosis of phakomatoses, only the most commonly encountered are considered here. The discussion is limited to cranial and intracranial manifestations. The cutaneous, spinal and visceral manifestations of phakomatoses are not described.
Neurofibromatosis type 1 (NF1)
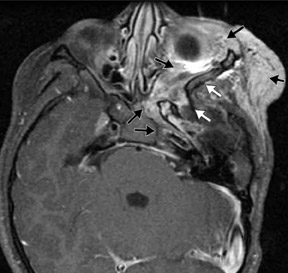
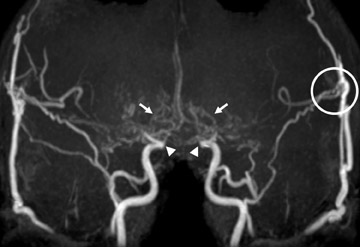
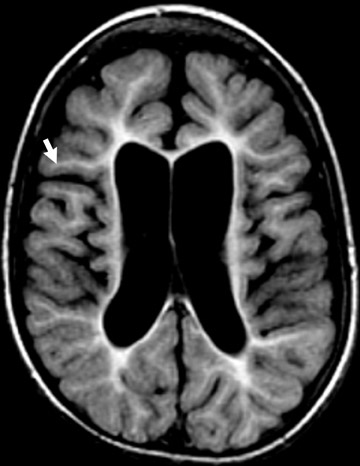
The genetic locus for NF1 is the long arm of chromosome 17 and inheritance is autosomal dominant. Neurological manifestations of NF1 are numerous. Most common among these is glioma of the optic pathway, which may be isolated to a single optic nerve, the optic chiasm or the entire optic pathway (Figure 1). Juvenile pilocytic astrocytomas are low-grade gliomas that may affect the optic pathway (Figure 1). Gliomas may also affect the brainstem, particularly the medulla, in patients with NF1, rather than localizing in the pons as occurs in the general population.
Magnetic resonance imaging (MRI) frequently reveals foci of white-matter signal abnormality on T2-weighted (T2W) sequencesin the basal ganglia, brainstem, cerebellar white matter and in the splenium of the corpus callosum. These are thought to be related to dysplastic formation of myelin, and they do not enhance—and usually regress by the late teenage years (Figure 1).1,2
The skull base, calvaria and orbit are commonly affected by dysplasia of the sphenoid bone. Sphenoid-wing dysplasia may allow herniation of the temporal lobe into the orbit and is commonly associated with plexiform neurofibroma of the orbit (Figure 1). Both ofthese orbital abnormalities can lead to exophthalmos. Plexiform neurofibromas and other neurofibromas are masses of Schwann cells, neurons and collagen that grow along the nerve of origin into the skull base and intracranially, where they cause mass effect on the brain parenchyma. Intracranial vascular dysplasia can result in stenoses that lead to development of moyamoya collaterals (Figure 1).1
Neurofibromatosis type 2 (NF2)
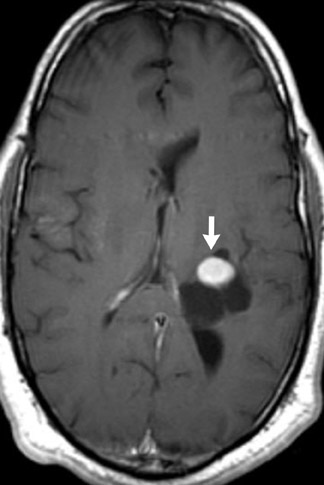
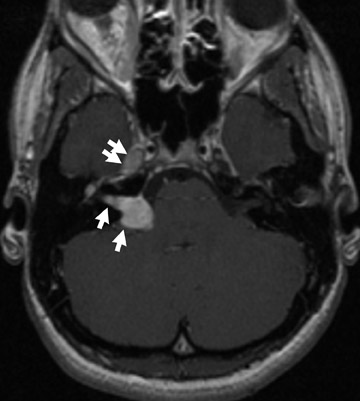
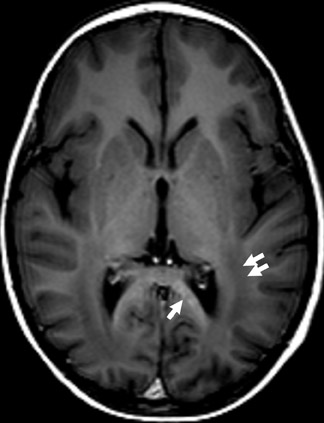
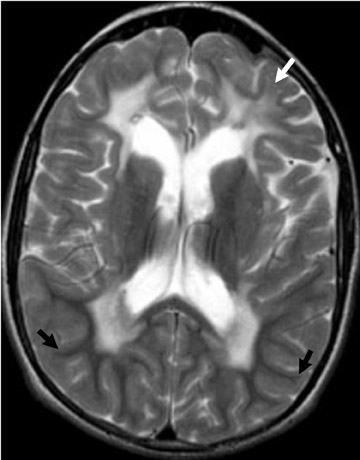
Chromosome 22 has been identified as the locus for the autosomal-dominant genetic abnormality in NF2. The quintessential neurologic finding of NF2 is bilateral vestibular nerve schwannomas. Schwannomas of other cranial nerves are not uncommon (Figure 2). Ophthalmologic findings in NF2 include cataracts, retinal hamartomas and optic nerve meningiomas.3
Several other tumors, neurofibromas, meningiomas, gliomas and schwannomas are found in patients with NF2. Meningiomas and schwannomas are often multiple (Figure 2).1
Sturge-Weber syndrome
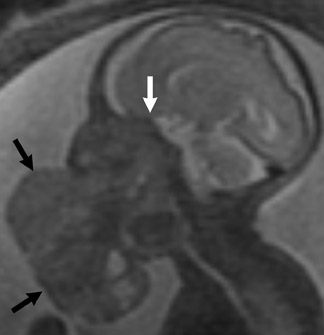
The inheritance pattern of Sturge-Weber is unknown, but familial cases are reported. The term encephalotrigeminal angiomatosis accurately describes the manifestations of Sturge-Weber syndrome that are the expression of a malformation of meningeal and facial vascular structures during weeks 4 to 8 of gestation. The intracranial vascular malformation or angiomatosis of the leptomeningeal vessels is a combination of arteries and capillaries that become fibrotic and subsequently calcify (Figure 3).1
Tuberous sclerosis
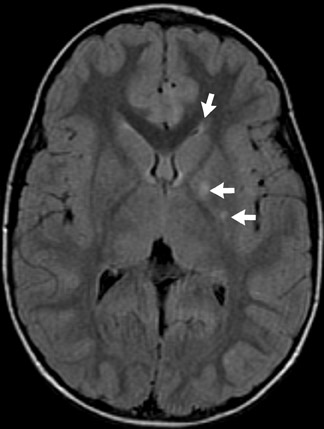
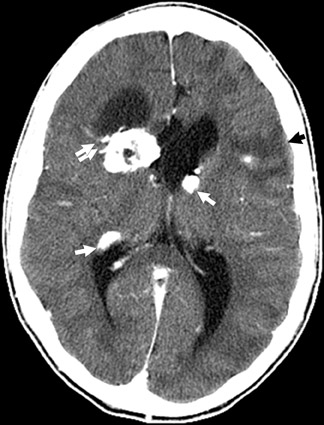
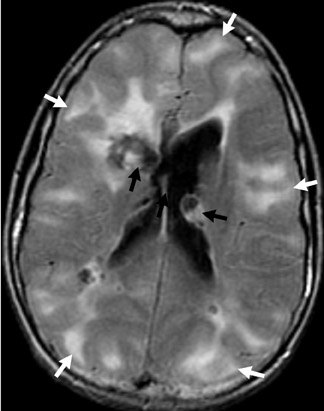
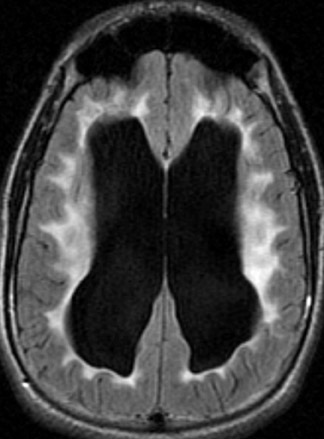
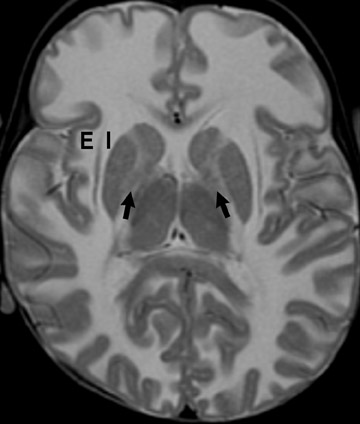
Tuberous sclerosis (TS) is inherited in an autosomal-dominant manner, or sporadically as a result of abnormal genes on chromosomes 9 and 16. The ensuing anomalies of neuronal proliferation and differentiation result in varied expression of hamartomas in the brain—referred to as subependymal nodules and cortical tubers. Nodules located adjacent to the foramina of Monro typically undergo degeneration to giant-cell neoplasms and subsequently grow large enough to obstruct the lateral ventricles, resulting in hydrocephalus. These may subsequently invade the brain parenchyma (Figure 4). Although historically called astrocytomas, these tumors are a mixture of glial- and neuronal-cell origin with giant cells.4
The subependymal nodules are usually calcified, making them quite conspicuous on computed tomography (CT) studies. MRI shows the protrusion of the nodules into the lateral ventricles. The nodules are slightly hyperintense on T1-weighted (T1W) sequences and enhance with gadolinium.
Hamartomas in the cerebral hemispheres consist of bizarre giant cells, fibrillary gliosis and disordered myelin. CT shows low attenuation in the subcortical white matter in broadened gyri, sometimes referred to as “empty gyri.” MR shows hyperintense T2 signal that can extend from the subcortical tissue through the centrum semiovale to the periventricular tissue (Figure 4).4
Inherited metabolic diseases and neurodegenerative disorders
Inherited metabolic diseases and neurodegenerative disorders can involve grey matter, white matter or both and lead to abnormal formation, breakdown, or loss of myelin. Dysmyelination (abnormal formation or production) and demyelination (breakdown or loss) can lead to abnormal neurodevelopment and/or degeneration, loss of neurodevelopmental milestones, seizures, and movement disorders. Numerous inherited dysmyelination disorders are known to be based on abnormalities of a specific cell organelle (lysosome, peroxisome and mitochondria) or due to deficiency or overproduction of enzymes or products of cellular metabolism.5
Myelination generally proceeds from inferior to superior, posterior to anterior and centrally to peripherally. MRI clearly reveals progression of myelination and can help suggest progression of dysmyelinating and demyelinating processes.
Disorders of myelination are usually not evident at birth because myelin is a relatively scarce ingredient in the brain of a newborn. Myelination is principally a postnatal process, with rapid proliferation during the first 2 years of postnatal life, and continuing throughearly adolescence.6 Therefore, when an infant or child fails to meet developmental milestones or loses milestones, disorders of myelination should be considered.
Although MRI and magnetic resonance spectroscopy (MRS) are helpful in suggesting a particular disorder, it remains crucial to confirm diagnoses with laboratory investigations in combination with clinical onset and progression of disease.
Metachromatic leukodystrphy (MLD)
MLD is inherited in an autosomal recessive manner. Genetic abnormalities on chromosomes 22 or 10 result in deficiencies in the lysosomal enzyme arylsulfatase A or prosapisine, respectively, which leads to myelin breakdown.5
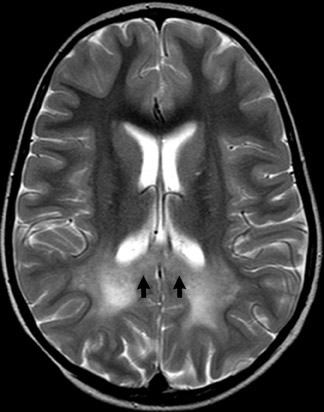
The demyelination is symmetric and spares subcortical U fibers early in the disease process. Periventricular T2 hyperintensity also involves the corpus callosum. Demyelination becomes confluent in the cerebral hemispheres but is less severe in the posterior fossa and spinal cord (Figure 5).7
Leigh’s syndrome
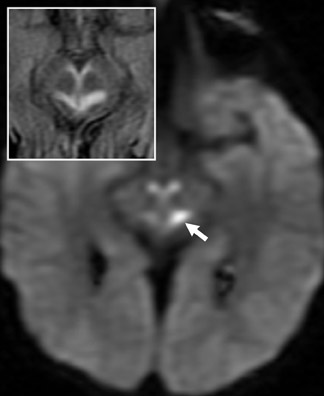
Impaired oxidative metabolism, resulting in reduced energy production in Leigh’s syndrome, can be X-linked or the result of several gene mutations that affect mitochondrial function.7,8 MR images show abnormally decreased T1 and increased T2 signal intensity in the caudate nuclei, putamina and globi pallidi (Figure 6).
MELAS syndrome
MELAS is an acronym for a disorder consisting of myopathy, encephalopathy, lactic acidosis and stroke. It is caused by a mutation in a mitochondrial gene.5,9
T2W images show wedge-shaped foci of hyperintensity in grey and white matter in any lobe of the brain. Lesions do not respect vascular territory distributions and may resolve on subsequent imaging studies. Imaging findings mimic vascular disease and stroke seen in elderly patients (Figure 7).7
Adrenoleukodystrophy
Autosomal-dominant, neonatal, X-linked and adrenomyeloneuropathic forms of adrenoleukodrystrophy are known. Each is a result of peroxisomal dysfunction which affects the brain and the adrenal glands.8
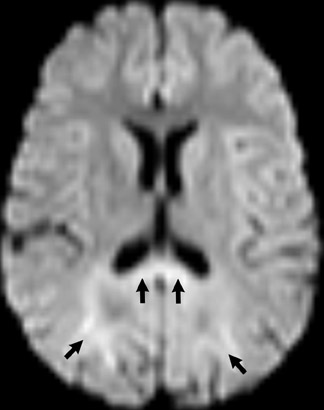
Imaging findings in the neonatal form are characteristic and suggestive, but not specific. Confluent high signal in the occipital white matter and involvement in the occipital lobes, through the splenium of the corpus callosum, suggests the X-linked form of adrenoleukodrystrophy. Diffusion, T2W and gadolinium-enhanced images show high signal at the margins, presumably in areas of active demyelination (Figure 8). Pathologic specimens correspond with the imaging findings. White matter involvement occurs from a posterior-to-anterior and central-to-peripheral manner, mimicking the normal maturation progression of myelination. Put another way, the demyelination process occurs in the same manner, or progression, as the progression of normal myelination.7,8
Pelizaeus-Merzbacher disease
Five types of leukodystrophy are classified as Pelizaeus-Merzbacher disease (PMD) or PMD-like disease, but are the result of different abnormalities of the proteolipidic protein 1 (PLP1) gene on the X chromosome, which is inherited in an X-linked, recessive manner.5
In this group of diseases formation and/or maturation of myelin is arrested prematurely, resulting in a pattern of hypomyelination on CT and MRI. Initial imaging studies may reveal an immature myelination pattern that does not progress to a more mature pattern on subsequent examinations. The corpus callosum may be thinned and diffuse white-matter volume loss and basal-gangliairon deposition is also seen (Figure 9).7,8
Alexander’s disease
Alexander’s disease (fibrinoid leukodystrophy) is a rare leukodystrophy in which a mutation of the gene for glial fibrillary acidic protein (GFAP) on chromosome 11 occurs due to autosomal-recessive inheritance with variable penetrance in male infants. Infantile, juvenile and adult forms of the disease are recognized.5,8
Low density in the frontal and parietal white matter, internal capsules and caudate heads on CT correlates with high T2 signal on MRthat progresses in a confluent manner from anterior to posterior (Figure 10).7,8
Canavan’s disease
Canavan’s disease (spongiform degeneration) is inherited in an autosomal recessive manner but may be sporadic, due to abnormality of the aspartylacylase (ASPA) gene on chromosome 17. Initially described in children of Ashkenazi Jewish descent, it may be seen in other populations as well.5
Imaging studies show abnormalities in grey and white matter. The cerebral white matter is diffusely abnormal, with confluent high signal in the subcortical white matter, internal and external capsules and the centrum semiovale. The globi pallidi are also characteristically involved,showing high signal on T2 (Figure 11).7,8
Congenital brain neoplasms
Congenital brain neoplasms are those discovered within the first 60 days of postnatal life.10 They are most commonly neuroepithelial and germ-cell tumors. Infants usually come to the attention of their parents or physicians because of increasing head circumference either because of the presence of the mass or resultant hydrocephalus. However, signs of increased intracranial pressure are rare given that the calvaria is capable of expanding without restraint via sutural widening.
Neonatal tumors are usually larger at the time of discovery, disproportionately malignant in histology and behavior, and carry a dismal prognosis. Tumors discovered in the prenatal period are more likely to be fatal.11,12
The recommended imaging modalities are brain sonography for infants <4 weeks of age, especially those with history of prematurity, and MRI for those >4 weeks. Sonography will allow an early identification of intracranial hemorrhage and hydrocephalus, both of which are more common than congenital neoplasm. The multiplanar capability of MRI allows the most productive evaluation of the brain, neoplasm and mass effect.
Teratoma
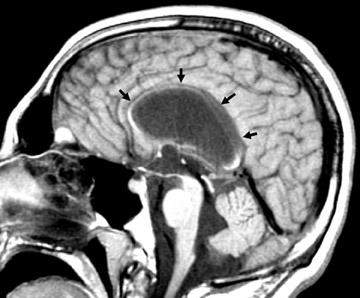
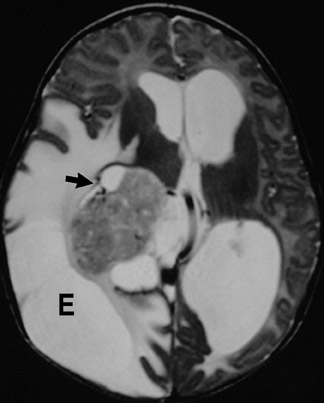
Arising from all 3 germinal layers, teratomas are heterogeneous masses that may be intracranial or extracranial. Intracranial teratomas tend to be supratentorial. Skull-base or extracranial tumors may or may not extend intra-cranially. Growth is along the path of least resistance.12
Imaging studies show a heterogeneous mass reflecting the presence of tissues from all 3 germinal layers and many contain calcification. Hydrocephalus may be a secondary finding (Figure 12).
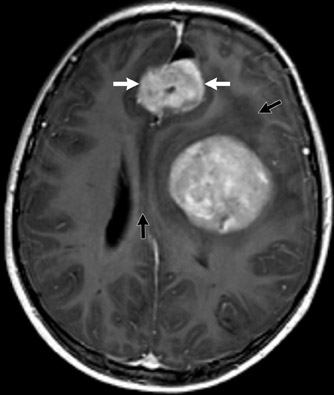
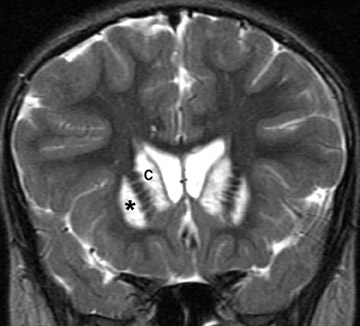
Astrocytoma
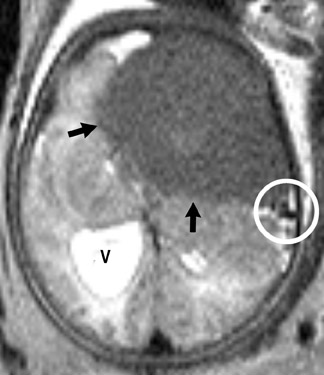
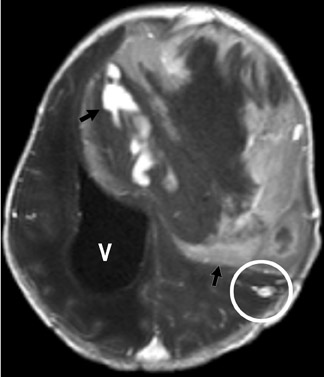
Astrocytoma is a tumor of the astrocytic line of glial cells of the brain or spinal cord. Fibrillary, infiltrative, pilocytic and anaplastic types are known.13
Imaging studies usually show a homogeneous mass and typically have a cystic component in the pilocytic form. Although hydrocephalus and increased head circumference are typically seen, they are not always present (Figure 13).
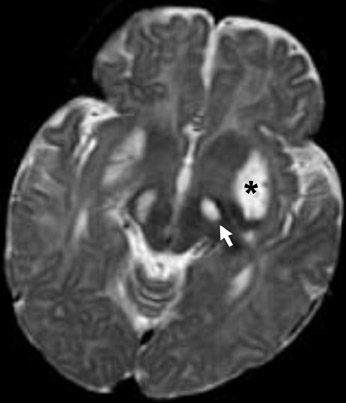
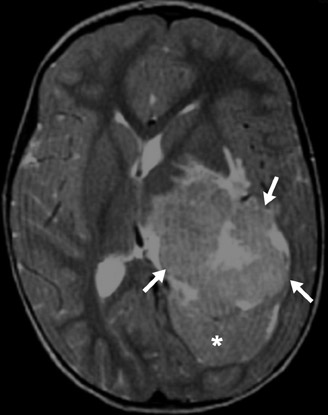
Choroid plexus papilloma and carcinoma
This intraventricular tumor leads to overproduction of CSF and subsequent hydrocephalus. Because it arises from the choroid plexus, the tumor is extremely vascular, leading to intense enhancement with contrast material. Vascular flow voids may be seen on MRI and small calcifications are best seen with CT. The carcinoma type of the choroid plexus neoplasm can invade brain parenchyma (Figure 14).10-12
Conclusion
Diagnoses in pediatric neuroradiology encompass a broad range of brain malformations, anomalies, inherited and metabolic disease processes. An understanding of basic brain embryology provides the basis for a more thorough understanding of these pathologic processes.
REFERENCES
- Barkovich AJ. The Phakomatoses. In: Barkovich AJ, ed., Pediatric Neuroimaging. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins;2005:440-505.
- Itoh T, Magnaldi S, White RM, et al. Neurofibromatosis type 1: The evolution of deep gray and white matter MR abnormalities. AJNR Am J Neuroradiol. 1994;15:1513-1519.
- Gareth D, Evans R. Neurofibromatosis type 2: In: Roach ES and Miller VS, eds. Neurocutaneous Syndromes. Cambridge, U.K.: Cambridge University Press;2004:50-59.
- Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004:63:1457-1461.
- Federico A, Rufa A, Battisti C, et al. Genetic leukoencephalopathies with unknown metabolic pathogenesis. Neurol Sci. 2001;22:S108-S112.
- Paus T, Zijdenbos A, Worsley K, et al. Structural maturation of neural pathways in children and adolescents: Invivostudy. Science. 1999; 283:1908-1911.
- Ruggieri PM. Metabolic, neurodegenerative and abnormal myelination disorders. In: WS Ball, Jr., ed. Pediatric Neuroradiology. Philadelphia, PA: Lippincott-Raven;1997:175-237.
- Barkovich AJ, Toxic and metabolic brain disorders. In: Barkovich AJ, ed. Pediatric Neuroimaging, 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins;2005:76-189.
- DiMauro S, Hirano M. MELAS. In: Gene Reviews October 2005 available online www.genetests.org. Accessed June 23, 2009.
- Ainstein LH, Boldery E, Naffziger H. A case report and survey of brain tumors during neonatal period. J Neurosurg. 1951;8:315-319.
- Buetow PC, Smirniotopoulous JG, Done S. Congenital brain tumors: A review of 45 cases. AJNR Am J Neuroradiol. 1990;11:793-799.
- Nejat F, Kazmi SS, Ardakani SB. Congenital brain tumors in a series of seven patients. Pediatric Neurosurgery. 2008;44:1-8.
- Stark AM, Modlich S, Claviez A, et al., Congenital diffuse anaplastic astrocytoma with ependymal and leptomeningeal spread: Case report. J Neurooncol. 2007;84:325-328.