Pediatric congenital heart disease
Images


Case summary
A full-term infant boy was delivered at 39-weeks gestational age to a 35-year-old woman via spontaneous vaginal delivery and then transferred to the Children’s Hospital due to a prenatal diagnosis of congenital heart disease. Delivery went well with no complications. Apgars were 8 at 1 min and 9 at 5 min. Physical examination showed a 2.9-kg infant with normal heart rate of 120 and respirations of 30. He was in no distress and was only slightly dusky in appearance, but not cyanotic. The baby had relatively high oxygen saturations at less than 1 hour of life (oxygen saturation of 92% on room air) and had been on room air since delivery. Physical examination was otherwise unremarkable.
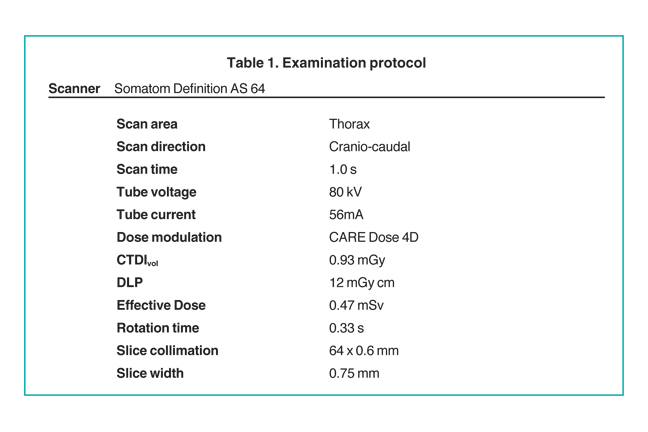
After delivery, an echocardiography was performed. A chest computed tomography (CT) scan was performed on day 5 of life prior to surgical repair of the heart disease. The study was performed on a Somatom Definition AS 64-row scanner (Siemens, Forscheim Germany). Images were obtained following administration of 6 mL of Optiray-320 nonionic contrast material (Covidien, St. Louis, MO). The acquisition parameters were 0.6-mm collimation, 56-auto mAs, 80-auto kV, a pitch of 1, and a 0.33-sec scan time. CT dose index (CTDI) volume was 0.93 mGy and dose length product (DLP) was 12m Gy·cm. Using the conversion factor (0.039 mSv/(mGy·cm))1 for chest examinations on day 1, effective dose (E) is estimated to be 0.47 mSv (12m Gy·cm x 0.039). On day 12 of life, the infant underwent repair of the congenital heart defect and repair of associated long-segment tracheal stenosis.
Imaging findings
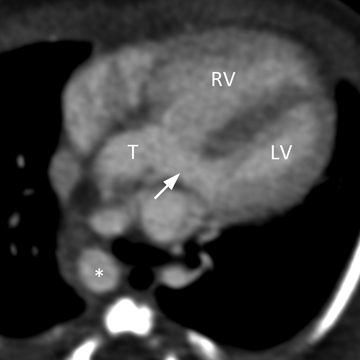
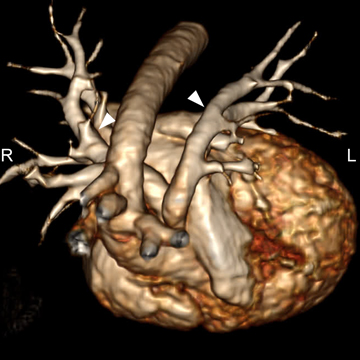
A common arterial trunk arises from both ventricles, which communicate via a large perimembranous ventricular septal defect (VSD). The arterial trunk divides into the aorta and main pulmonary artery, and the main pulmonary artery subsequently divides into left and right branch pulmonary arteries. (Figure 1 A to D).
Diagnosis
Truncus arteriosus with ventricular septal defect (Collett and Edwards type I; Van Praagh type 1A).
Discussion
Truncus arteriosus is a congenital heart disease resulting from failure of truncoconal septation. It occurs in 1% to 2% of infants with congenital heart defects.2 Associated genetic abnormalities include chromosome 22q11 deletion, trisomy 8, and chromosomal 10p deletion. Truncus arteriosus is considered a cyanotic heart disease due to the admixture of pulmonary and systemic venous blood, but cyanosis is often not a dominant feature unless there is associated pulmonary artery stenosis. Affected patients present with findings of heart failure soon after birth.
In this malformation, a single arterial trunk with a single valve arises from the heart giving rise to the pulmonary, systemic, and coronary arteries. In virtually all patients, there is a ventricular septal defect (VSD) and a single semilunar valve. The single semilunar valve may contain 2 or more leaflets, which are often dysplastic and associated with truncal insufficiency or stenosis. A tricuspid truncal valve is the most common type (69% of cases). In most instances, the semilunar valve overrides the true interventricular septum, and thus, both ventricles. Among the associated aortic anomalies are right aortic arch (25% to 33% of cases), aortic arch interruption (11% to 38% of cases), and single coronary artery (10% to 20% of cases).2
Two principle classification systems for truncus arteriosus exist. The classification scheme of Collett and Edwards bases nomenclature upon the sites of origin of the pulmonary arteries.3 In type I, a common arterial trunk divides into an aorta and short pulmonary trunk, which then divides into left and right pulmonary arteries (seen in this patient). In type II, the left and right branch pulmonary arteries have separate origins from the posterior aspect of the truncus. In type III, the left and right pulmonary arteries arise from the sides of the truncus, and in type IV the pulmonary arteries arise from the descending aorta. Often referred to as pseudo-truncus, this is currently not thought to be a type of truncus arteriosus but rather to be a form of pulmonary atresia with VSD (tetralogy of Fallot with pulmonary atresia).
The Van Praagh classification takes into consideration the presence of a patent ductus arteriosus and interrupted aortic arch, associated anomalies that have important surgical implications.4 The type 1 anomaly is similar to the type I truncus in the Edwards and Collett classification. In type 2, the right and left branch pulmonary arteries arise from the common arterial trunk. Their proximity to one another is not specified. In type 3, there is the absence of 1 branch of the pulmonary artery from the common arterial trunk, and either ductal or aortic origin of that pulmonary artery. In type 4, the aortic arch is hypoplastic or interrupted, and there is a large patient ductus arteriosus. The Van Praagh classification also specifies the presence of a VSD (type A), or the absence of a VSD (type B). Thus, each case of truncus arteriosus has a nomenclature that includes a letter and a number.
Currently, surgical management consists of complete primary repair, with closure of ventricular septal defect, detachment of the pulmonary arteries from the aorta with reconstruction of the right ventricular outflow tract and commitment of the common arterial trunk to the left ventricle. In effect, the common trunk becomes the ascending aorta and the truncal valve is the aortic valve.2
Conclusion
We report a case of truncus arteriosus. CT provides excellent characterization of anatomy and may be especially useful in defining complex pulmonary arterial anatomy or arch anomalies for surgical planning.5 CT scanners can provide levels of anatomic detail with very low radiation dose to the baby. Survivors of truncus arteriosus undergo complete surgical repair.
References
- Shrimpton PC, Hillier MC, Lewis MA, Dunn M. National survey of doses from CT in the UK: 2003. Br J Radiol. 2006;79:968-980.
- Connelly M. Common arterial trunk. In: Gatzoulis MA, Webb GD, Daubeney PEF. Adult Congenital Heart Disease. Edinburgh: Churchill Livingstone. 2003;265-271.
- Collett RW, Edwards JE. Persistent truncus arteriosus: A classification according to anatomic types. Surg Clin North Am. 1949;29:1245-1270.
- Van Praagh R, Van Praagh S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. A study of 57 necropsy cases. Am J Cardiol. 1965:406-425.
- Siegel MJ. Heart. In Siegel MJ, ed. Pediatric Body CT. Philadelphia, PA: Lippincott Williams & Wilkins. 2008;145-175.