Pediatric chest: A review of the must-know diagnoses
Images








Diagnostic imaging of the pediatric chest patient differs vastly from that of the adult patient. Both common and rare entities may manifest differently in younger children than in adults. Additionally, the sort of pathology encountered in pediatric patients also varies. Identifying the range of pediatric thoracic abnormalities on various diagnostic modalities is essential to providing optimal care for our pediatric patients. Referring clinicians look to radiologists to provide diagnoses that will direct clinical decision-making. With these aims, this review will cover the “must-know” congenital and acquired lesions in the pediatric chest.
Congenital parenchymal lesions
While congenital lung lesions may not be routinely encountered in daily imaging, they are common considerations in diagnostic pediatric radiology. Early recognition is crucial to prevent misdiagnosis and mismanagement.
Congenital thoracic lesions are thought to originate from disturbances during the embryogenesis of lung and airway development. The imaging appearances of these lesions often demonstrates significant overlap. In fact, even on histopathologic analysis, these lesions may share similar characteristics, as some are truly hybrid entities. In current radiologic and pathologic analyses, congenital lung lesions are regarded as a continuous spectrum of developmental abnormalities, from cystic airway lesions to mixed cystic airway/vascular lesions to purely vascular lesions.
As our understanding evolves, so do the nomenclature and classification of these entities. Under this umbrella, the most commonly described congenital chest lesions are congenital pulmonary airway malformations (CPAM), bronchopulmonary sequestrations (BPS), bronchogenic cysts, and congenital lobar overinflation (CLO). Among these, CPAM and BPS most commonly present as hybrid lesions. These lesions are now routinely detected on fetal ultrasounds and are often identified prior to birth. While the appearance of congenial lung masses on fetal imaging is beyond the scope of this paper, most of these entities present as hyperechoic, space occupying masses, which shift the mediastinal contents away from the involved hemithorax on fetal sonography.
Congenital pulmonary airway malformations
Formerly termed congenital cystic adenomatoid malformation, this lesion is now more aptly called congenital pulmonary airway malformation (CPAM) to encompass the gamut of histological appearances. CPAM is the most commonly encountered congenital cystic lesion,1 thought to result from abnormal branching of immature bronchioles during lung development. The incidence is likely underestimated at 1 in 25,000 to 1 in 35,000 live births.2
CPAMs are hamartomatous anomalies that communicate with the tracheobronchial tree.3 Histologically, these lesions look like terminal bronchioles lined with respiratory epithelium, lacking cartilage or bronchial glands.1 The exact insult causing this abnormality and the precise timing of its development are unknown; it may form anywhere between 5-22 weeks gestation. Pure, non-hybrid, CPAMs have normal arterial and venous connections to the pulmonary circulation. Hybrid CPAMs that occur concomitantly with abnormal vascular connections feeding or draining the lung lesions may occur alongside additional abnormalities, including congenital heart disease and neural tube defects. Not surprisingly, patients with these additional associations have a higher incidence of stillborn birth.4
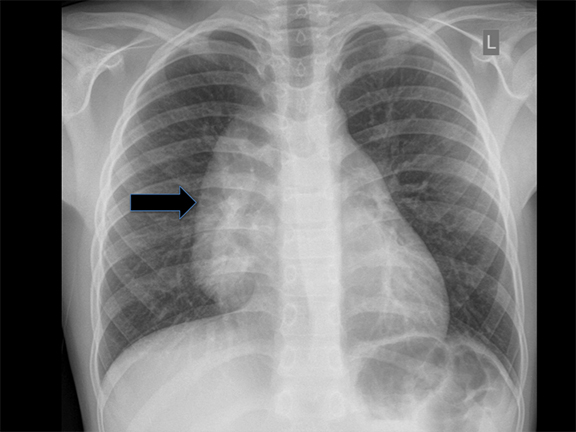
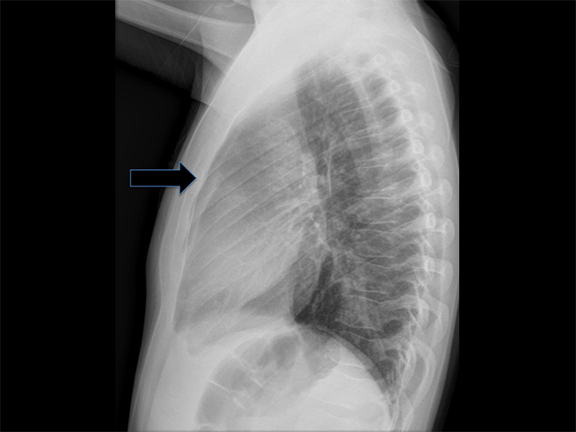
CPAMs may contain larger cysts greater than 2 cm, smaller cysts between 0.5 and 2 cm, or microcysts, which may create the appearance of a solid mass on imaging. The vast majority of these lesions are solitary, with a lower lobe predilection.5 When bilateral CPAMs are present, there is a higher risk of congenital high airway obstruction syndrome (CHAOS).5 CPAMs often present with respiratory symptoms including tachypnea, retractions, and cyanosis within first month of life.4,56 Fewer than 10% present after one year of age.5 Later presentations include recurrent or persistent pneumonia (most common), lung abscess, pneumothorax, reactive airway disease or, rarely, malignant transformation. Some CPAMs may regress, especially the microcystic type.2
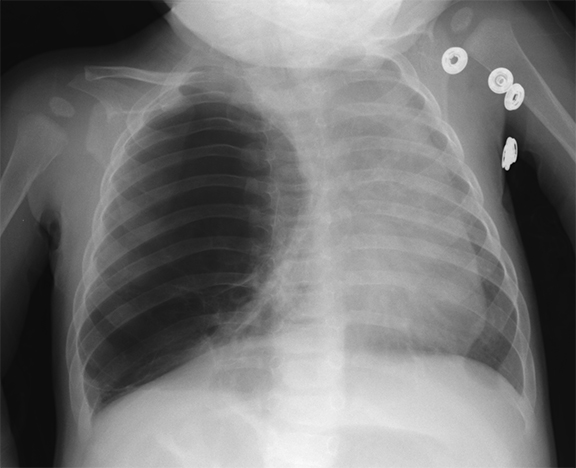
On radiographs, CPAMs are hyperlucent and multicystic, with occasional solid components. Large lesions may cause lung hyperexpansion with mediastinal shift or flattening of the hemidiaphragm (Figure 1). Air-fluid levels may be seen secondary to residual fluid in the initial postnatal period. In older patients, air-fluid levels typically reflect the sequelae of resolving infection. When older patients present with infected CPAMs, it is important to differentiate these lesions from necrotizing pneumonia, with findings including hyperexpanded lungs and a lack of air bronchograms favoring an infected CPAM. Computed tomography angiography (CTA) is essential in distinguishing these lesions from other congenital lung lesions, as the presence of a systemic feeding vessel may herald a hybrid lesion or sequestration. This vessel must be recognized prior to surgical intervention.
Treatment of CPAMs involves surgical resection, either via lobectomy or segmental resection, if multiple lesions are present.2 Early surgery prevents infection and reduces the rarer risk of malignancy, which has been identified as early as 13 months of age.7 Associated malignancies include embryonal rhabdomyosarcoma, pleuropulmonary blastoma and bronchoalveolar carcinoma.8-10
Bronchopulmonary sequestrations
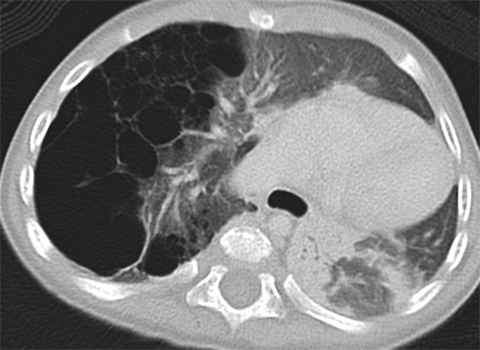
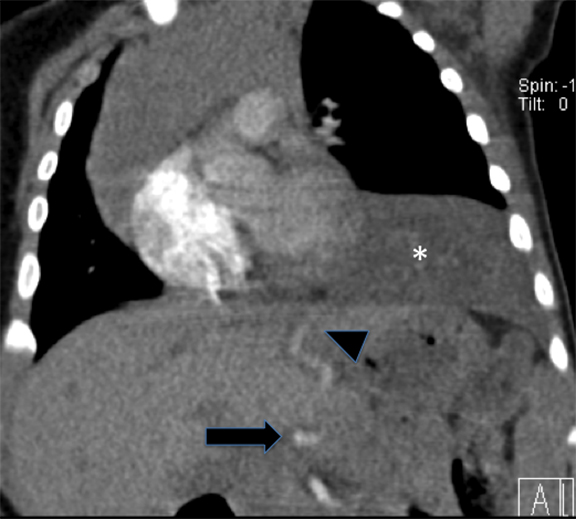
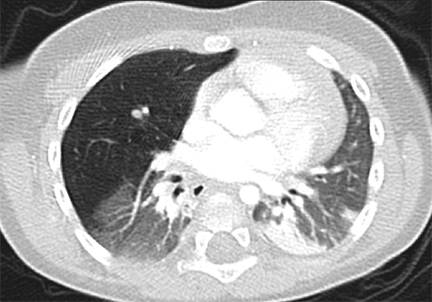
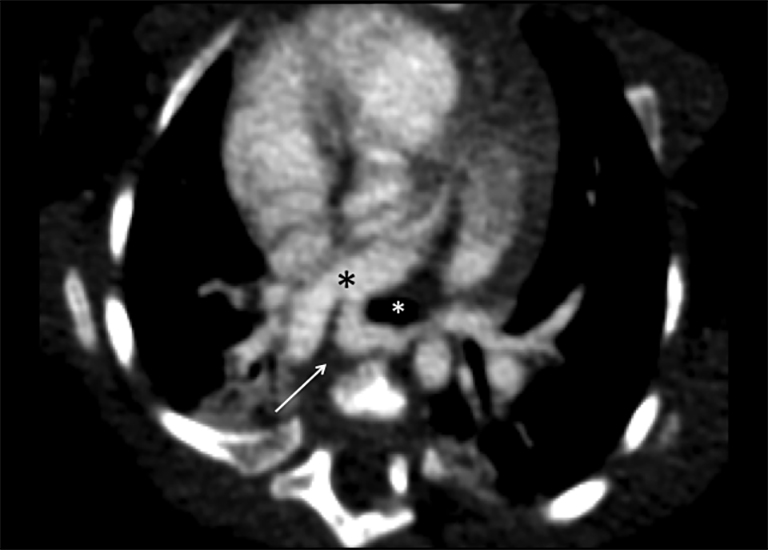
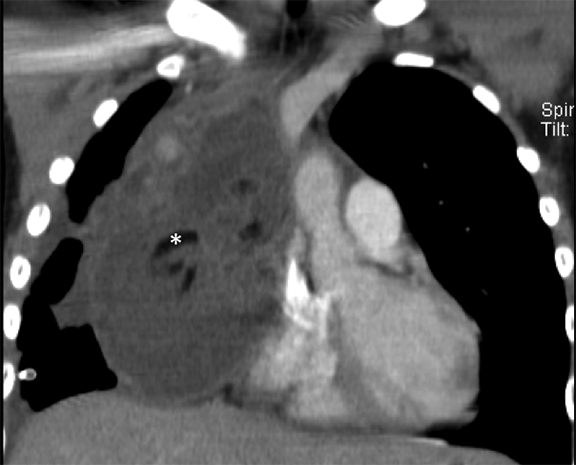
Bronchopulmonary sequestrations (BPS) are composed of nonfunctioning pulmonary tissue that lacks communication with the tracheobronchial tree, possibly originating from a supernumerary lung bud.5 These lesions characteristically have a systemic arterial supply arising from vessels including the thoracoabdominal aorta, splanchnic vessels, or rarely, coronary arteries (Figure 2). Sequestrations may be asymptomatic or present with respiratory distress, infection, feeding difficulties or even, rarely, congestive heart failure.11
There are two commonly described subtypes of BPS: extralobar and intralobar (Table 1). Extralobar sequestrations are discrete and invested in their own separate pleura. They are typically located in the left lower chest, but can be also be within or below diaphragm. Typically, these lesions demonstrate systemic venous drainage to the superior vena cava or azygos veins. In some case, extralobar sequestrations may be associated with other congenital abnormalities, including congenital diaphragmatic hernias and CPAMs.5 Intralobar sequestrations are closely associated with adjacent lung, demonstrate pulmonary venous drainage, and are generally isolated without other associations. This subtype presents in later childhood.
Sequestrations may sometimes be visible in neonatal period via ultrasound, as well-defined, homogeneous, isoechoic masses. Color Doppler may demonstrate the pathognomonic systemic blood supply. Presence of concurrent cysts may suggest a hybrid lesion. The two subtypes of sequestration cannot be reliably distinguished on imaging unless a pleural effusion surrounds the extralobar sequestration or if the sequestration occurs in upper abdomen and is, by definition, extralobar. An upper abdominal extralobar sequestration must be distinguished from a suprarenal neuroblastoma, which lacks systemic arterial supply and demonstrates metaiodobenzylguanidine (MIBG) uptake on nuclear scintigraphy.
On CT scans, sequestrations may be homogeneous or heterogeneous masses, sometimes with cystic changes. Characteristically, these are solid lesions surrounded by peripheral emphysematous changes. MRI/MRA chest may also be performed to assess these lesions. However, CT is generally preferred due to better resolution of lung parenchymal findings.
Some sequestrations regress postnatally, but symptomatic infants are managed surgically.2 Asymptomatic lesions are should be adequately characterized by cross-sectional imaging to define the feeding vessel then electively resected in order to minimize risk of infection or hemorrhage.
Congenital lobar overinflation
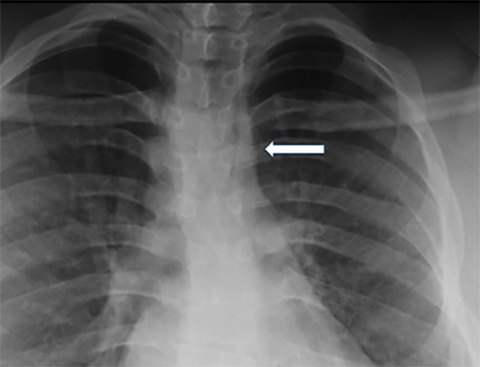
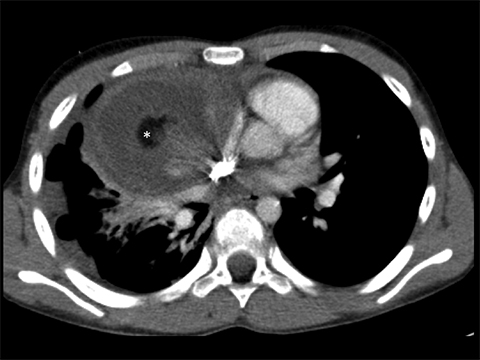
Congenital lobar overinflation (CLO) was previously termed “congenital lobar emphysema”—a misnomer, since these lesions lack the destruction of alveolar walls that typically denotes ‘emphysema’. Typically, CLO affects a single lobe of the lung, although multiple lobes or segments within a lobe may be affected. In decreasing frequency, the most commonly affected lobes are: left upper, right middle and right upper lobes.12 CLO arises due to narrowing of the airway lumen, followed by subsequent obstruction from various etiologies such as endobronchial lesions (e.g. mucosal webs), extrinsic compression from vascular anomalies or congenital deficiency of bronchial cartilage and ensuing bronchomalacia.13 The obstructed region becomes hyperinflated from collateral drift through check-valve mechanism at the level of the bronchus, with a greater amount of air entering on inspiration than exiting on expiration. CLO is sometimes associated with congenital heart disease, namely, patent ductus arteriosus (PDA) and ventricular septal defect (VSD).12
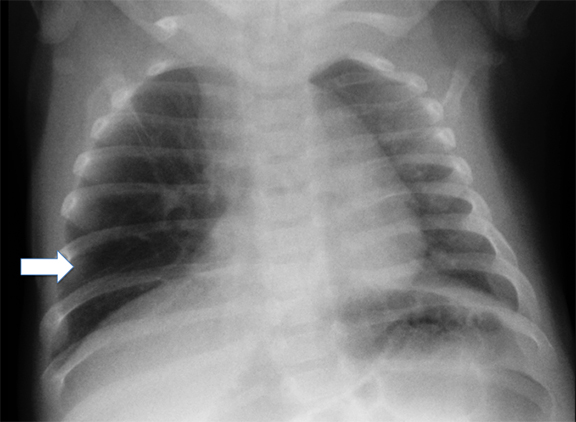
Diagnosis is made via radiography and computed tomography (CT). CLOs tend to be symptomatic lesions, presenting with respiratory distress in early life. At birth, these lesions may be may be fluid-filled and radiopaque. As fluid is reabsorbed, lesions may first demonstrate interstitial reticulation and, finally, become hyperlucent once significant air trapping occurs. There may be associated mass effect with contralateral atelectasis, flattening of hemidiaphgram and increased rib spacing in affected lobe (Figure 3). Vessels will be attenuated, and CLO must be differentiated from pneumothoraces which demonstrate a pleural line and lack bronchovascular markings. Most lesions are treated via lobectomy to allow normal lung development and symptomatic relief.14
Bronchial atresia
Unlike the previously described thoracic abnormalities, bronchial atresia is rarely detected prenatally. Most often, these are discovered due to recurrent symptoms or identified incidentally. Bronchial atresia occurs due to obliteration of a focal segment of bronchus, with normal development of the more distal lung.15 Most commonly, this affects bronchial structures at the level of the segmental/subsegmental bronchus.15 If bronchial atresia occurs centrally, at the level of the mainstem bronchus, it is usually lethal in utero or the immediate postnatal period.
Peripheral bronchial atresias present in older children due to recurrent infections. Radiographs may demonstrate an overinflated portion of lung. A bronchocele may be identified distal to the atretic bronchus, appearing as a fluid-filled, tubular, branching structure, finding which is considered pathognomonic for bronchial atresias. Symptomatic atresias that serve as a nidus for recurrent infections are surgically removed. Incidental, asymptomatic atresias may not require intervention.
Vascular rings
Vascular rings arise from the persistence of embryonic aortic arches.
Double aortic arch
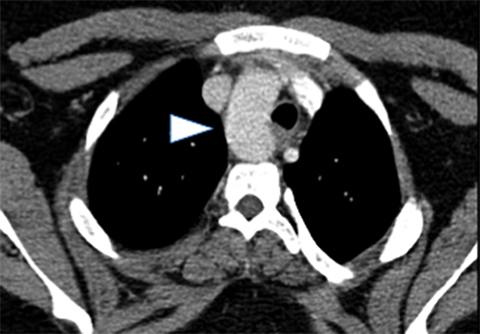
Double aortic arches account for roughly 50% of symptomatic vascular rings.16 These vascular abnormalities occur when both the right and left fourth embryonic aortic arches persist, encircling both the trachea and esophagus. While both arches may be patent, a segment of one arch may also be hypoplastic or atretric. Most patients demonstrate dominant right aortic arches, which are larger and rostral to the left arch (Figure 4).
Characteristically, tracheal compression from the vascular ring produces inspiratory stridor that worsens during food ingestion. Associated tracheomalacia may worsen these symptoms. On fluoroscopic barium swallow studies, bilateral indentations may be visible on the upper esophagus, with additional compression along the posterior esophageal wall. Cross-sectional imaging findings include the “four vessel sign,” created when each arch gives rise to its ipsilateral subclavian and common carotid arteries. Treatment involves surgical ligation of the smaller arch.
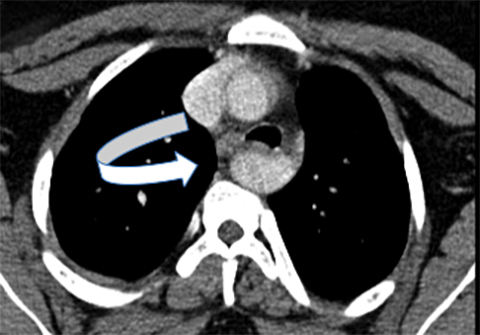
Right aortic arch
A right aortic arch reflects persistence of the right fourth embryonic arch. A right arch will cross over the right mainstem bronchus. The location of the descending aorta is variable; along either the right or left posterior mediastinum (Figure 5). Two main subtypes of right aortic arch are: a right aortic arch with aberrant left subclavian artery (RLSCA) or a right arch with mirror-branch type pattern. The RLSCA subtype is asymptomatic in up to 95% of cases. A complete ring is only formed if a left ligamentum arteriosum is present, which may cause respiratory symptoms.16 The aberrant left subclavian artery may have aneurysmal dilatation at its origin, arising from a remnant of the left dorsal aortic root. This is termed the “diverticulum of Kommerell”. If vascular rings are symptomatic, they are treated by transection of the ligamentum arteriosum.
The right aortic arch with mirror branching is the second-most common right aortic arch branching pattern and reflects a mirror image of the left aortic arch.17 Without a true vascular ring, this branching pattern is largely asymptomatic. However, it is notable for a high association with congenital heart disease, including Tetralogy of Fallot and truncus arteriosus. The first branch of the mirror arch is the left brachiocephalic artery, followed by the right common carotid artery and the right subclavian artery.
Left aortic arch with aberrant right subclavian artery
The left aortic arch with aberrant right subclavian artery (LRSCA) is the most common congenital aortic arch anomaly, occurring in roughly 1 of every 200 people.17 Since this vascular branching pattern is not associated with respiratory compromise, it is typically identified incidentally. When symptomatic, it is usually due to mass effect on the posterior esophagus causing “dysphagia lusoria” or difficulty swallowing. Chest x-rays may be normal or demonstrate subtle mediastinal widening. If a barium swallow is performed, an oblique region of mass effect may be seen along the course of the aberrant vessel as it compresses the posterior esophagus, extending superiorly from left to right. In symptomatic patients, surgical ligation of right ductus may be performed.
Anomalous origin of left pulmonary artery
Commonly termed “pulmonary sling,” this anomaly occurs when the left main pulmonary artery arises from the right main pulmonary artery rather than from the pulmonary trunk (Figure 6). The anomalous pulmonary artery traverses between the trachea and esophagus, producing mass effect along the anterior esophagus and the posterior trachea. Patients with this anomaly are often symptomatic, presenting with stridor, hypoxia, and often asymmetric lung aeration. There may be concomitant tracheal rings or tracheomalacia.
Mediastinal masses
Pediatric mediastinal masses are best approached by considering the types of masses that may occur in each mediastinal compartment.
Anterior mediastinum
The most common anterior mediastinal mass in children is lymphoma. Lymphomas are further classified into Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (NHL). Both may present with nonspecific symptoms associated with mass effect on the mediastinal structures, including cough, dyspnea, dysphagia, SVC syndrome or hemoptysis. Lymphoma may also manifest with additional “B symptoms:” fever, night sweats, and weight loss.
At imaging, both HL and NHL can present as an enlarged conglomeration of intrathoracic lymph nodes. Roughly 85% of HL present within intrathoracic involvement, usually as an anterior mediastinal mass. On chest radiography, this may appear as mediastinal widening, displacement of the trachea, and loss of the retrosternal clear space. Less commonly, HL will present with pulmonary nodules, consolidation and pleural effusions.16 In contrast, only about 50% of NHL demonstrates enlarged intrathoracic lymph nodes. Pulmonary findings in NHL are more diverse and may range from pulmonary nodules (which may cavitate), consolidation to interstitial thickening. Cross-sectional CT imaging of chest and body is essential for staging. Treatment generally involves chemotherapy and/or radiation, which may be started urgently in cases of airway or vascular compromise.
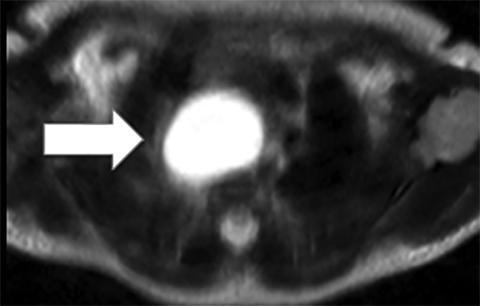
Intrathoracic germ cell tumors (GCT) arise following the arrested migration of primitive germ cells that remain in the anterior mediastinum rather than descending into the gonads. These tumors share similarities with primary tumors arising from the ovaries and testes. GCTs account for approximately 20% of pediatric mediastinal tumors.18,19 Approximately 15% are malignant, which portends poor prognosis.20
Teratomas represent a subtype of GCT, comprising approximately 60% of all mediastinal germ cell tumors. If a child presents with calcified mediastinal mass, this is assumed to represent a teratoma until proven otherwise. Teratomas may be mature and benign or immature with malignant potential; imaging alone may not be able to determine whether a teratoma is malignant or benign. Cross-sectional imaging will often demonstrate a mass comprised of various densities, including fat, fluid and calcification. The presence of fat is virtually diagnostic of a teratoma (Figure 7).
GCT and teratomas are usually asymptomatic until they are large, at which point they may cause respiratory distress and compromise the airway. Once they are detected in the mediastinum, the pelvis should also be assessed to exclude a primary gonadal tumor. Since malignant teratomas are often quite large, patients may require preoperative chemotherapy prior to surgical resection.18,20
Thymomas are infrequent in the pediatric population comprising only 1-2% of mediastinal masses in children.16 As in adults, thymomas many present with paraneoplastic syndromes including myasthenia gravis, hypogammaglobulinemia or red cell aplasia.
Middle mediastinum
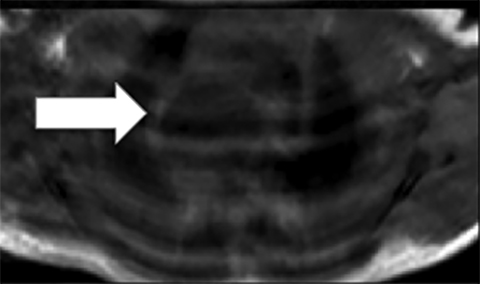
The most common middle mediastinal masses in children are foregut duplication cysts. Mediastinal foregut duplication cysts are further classified into bronchogenic, esophageal and neurenteric cysts.
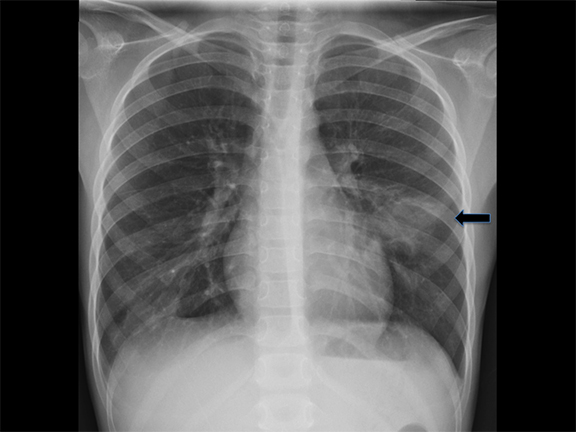
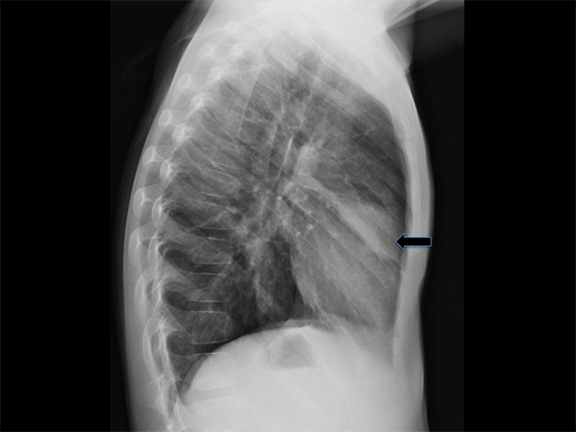
Bronchogenic cysts arise due to abnormal lung budding during ventral foregut development during the first trimester.16 Most arise near the carina and right paratracheal region (Figure 8), though up to 20% are contained within lung parenchyma. Histologic examination reveals respiratory epithelial lining.
Esophageal duplication cysts result from abnormal development of the posterior portion of the embryonic foregut. These often present with dysphagia and are commonly associated with the upper third of the esophagus. In rare cases, migration with the lung bud may lead to an intraparenchymal location. Histology reveals gut epithelia including submucosal and muscular layers.
Although neurenteric cysts are usually found in the posterior mediastinum, these lesions are typically classified with other foregut cysts and will be briefly covered here. Neurenteric cysts result when the neural crest and gastrointestinal tract fail to separate. These cysts may be associated with osseous abnormalities of the adjacent spine and will contain both GI and neural histologic elements.
On imaging, each of the foregut cysts appears as a fluid-filled, well-circumscribed round or oval mass. CT may demonstrate a lesion of water attenuation or slightly higher, depending on the amount of internal proteinaceous contents. No intralesional enhancement is seen, though peripheral enhancement may be present in the setting of infection/inflammation. Treatment is generally surgical resection, especially for symptomatic lesions.
Diffuse middle mediastinal adenopathy is generally due to lymphoma. However, unilaterally enlarged hilar lymph nodes are suggestive of infections, particularly, tuberculosis. Metastatic mediastinal adenopathy can also be considered in the correct clinical setting. If lymph nodes are calcified, osteosarcoma or treated lymphoma should be on the differential.
Posterior mediastinum
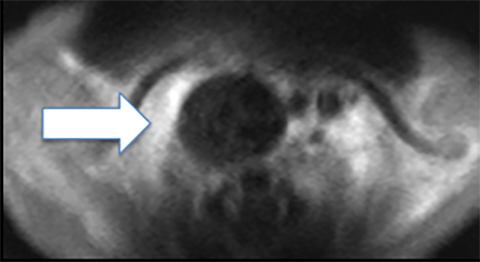
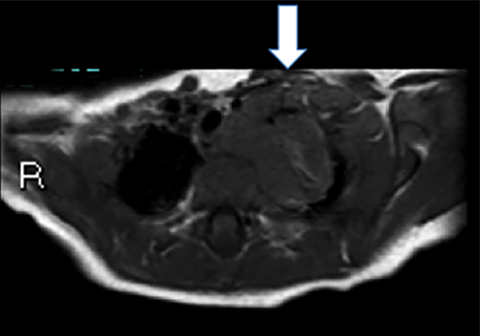
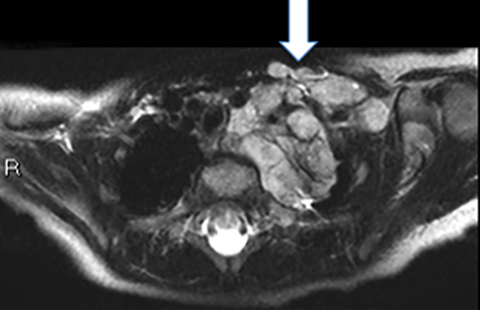
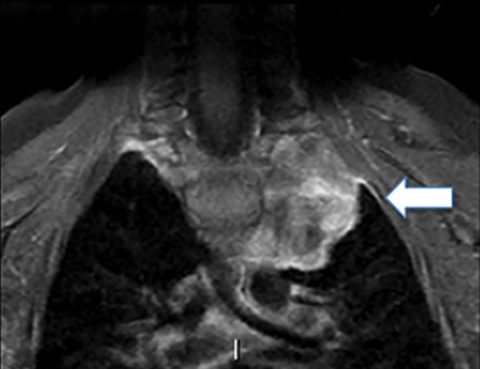
As discussed above, neurenteric cysts should be strongly considered when a cystic mass is encountered in the posterior mediastinum. When a solid mass is identified, neurogenic tumors associated with the sympathetic chain should be high on the differential. These tumors exist on a spectrum, ranging from malignant neuroblastoma to ganglioneuroblastoma and benign ganglioneuroma. Of these entities, neuroblastoma is the most common, and represents the most common extra-cranial solid tumor of childhood.
Neuroblastomas arise from primitive neural crest cells. While most commonly seen in the adrenal gland, neuroblastomas also frequently arise in the posterior mediastinum. Systemic symptoms include irritiability, fever, weight loss and anemia. Cord symptoms such as bladder/bowel dysfunction or lower extremity weakness should suggest extension into the spinal canal or neural foramen.
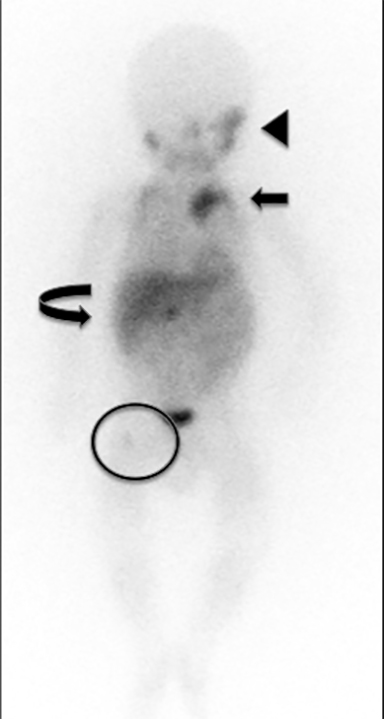
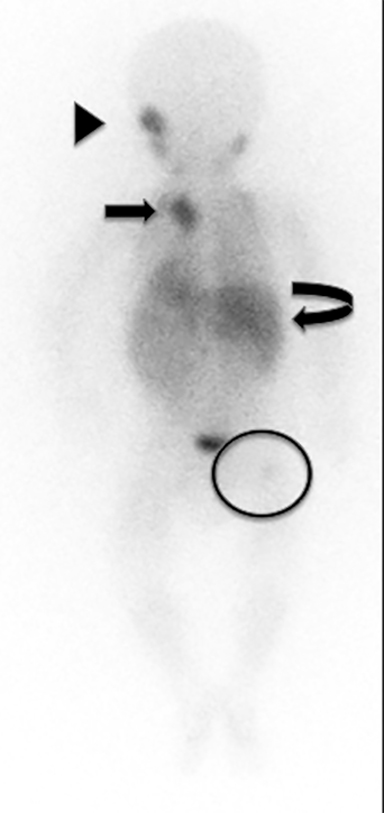
Chest radiography may demonstrate abnormal paravertebral opacity with associated widening of intercostal spaces or rib erosions. CT or MRI scans may demonstrate intralesional calcification and extension into the neural foramen. Neuroblastomas are invasive and often encase vessels. These tumors may be vascular and demonstrate robust, albeit heterogeneous, enhancement. Due to neural origins and catecholamine production, nuclear scintigraphy using metaiodobenzylguanidine (MIBG) aids in detecting extent of tumor and distant metastases (Figure 9). Typically, thoracic neuroblastomas have a more favorable outcome than abdominal neuroblastomas.
Ganglioneuroblastomas are intermediate tumors with malignant potential that may demonstrate distant metastases, while ganglioneuromas are benign. Both tumor types tend to be more fusiform in appearance and smaller than neuroblastomas, as well as less invasive. These more mature tumors are also seen in slightly older children. Biopsy is required for conclusive diagnosis.
Round pneumonia
Round pneumonia is a phenomenon that typically occurs in children due to undeveloped communicating passages in the air spaces called pores of Kohn and channels of Lambert.21 Underdevelopment of these channels allows for centrifugal spread of fluid and/or bacteria leading to rounded appearance of infection. Round pneumonias are usually pneumococcal infections in children under the age of 8, presenting as solitary, well-defined, lesions in the posterior and lower lobes (Figure 10).22 Air bronchograms may be present. At imaging, round pneumonias may appear similar to a parenchymal bronchogenic cysts, pleuropulmonary blastoma or metastatic disease. However, a clinical history of acute/subacute onset of symptoms, with fever, leukocytosis, and cough should provide the diagnosis. Round pneumonias are treated with antibiotics and rarely progress to lobar consolidation.
Chest wall
Ewing sarcoma
Palpable chest wall masses and rib anomalies typically include benign diagnoses including fibrous dysplasia and chronic recurrent multifocal osteomyelitis. However, the most worrisome and common malignant tumor of the chest wall is Ewing sarcoma and related tumors, including primitive neuroectodermal tumor (PNET) and Askin tumor of the chest wall.23,24
Ewing sarcoma is most frequently encountered in the first 3 decades of life, peaking between 10-15 years of age, with slight male predilection. 95% of patients are of Caucasian descent.25 A translocation on the long arms of chromosomes 11 and 22 (t[11;22]) is seen in up to 90% of these tumors.24,26 Many also express CD99 in the tumor membrane.27
Chest radiography may demonstrate pleural involvement including fluid and/or direct tumor extension. Pulmonary parenchymal disease and enlarged mediastinal lymph nodes may also be seen. Similar to other aggressive osseous tumors, bony manifestations include permeative, moth eaten lucencies, a wide zone of transition and associated soft-tissue mass. Periosteal reaction may be lamellated or spiculated. Smaller intra-osseous sclerotic components can be seen in flat bones, including the ribs.
Cross-sectional imaging allows for the assessment of cortical destruction and aids in defining the extent of the soft tissue mass. The superior soft tissue contrast of MRI renders it ideal for evaluating Ewing sarcoma. On T1-weighted sequences, tumors tend to be homogeneous, of intermediate intensity while on T2-weighted sequences, tumors are homogenous with low-to-intermediate signal intensity. These signal characteristics are attributed to high cellularity of Ewing sarcomas. A heterogeneous appearance on MRI may reflect intratumoral hemorrhage and/or necrosis. When posterior ribs are involved, MRI of spine should also be performed to assess for tumor extension.
Non-accidental trauma
In children under 2 years of age, fractures are highly associated with physical abuse.28,29 Some of these non-accidental injuries manifest in the bony thorax and may be incidentally discovered on chest radiographs. For instance, posterior rib fractures occurring at the head/neck of the rib near the costovertebral articulation, represent one of the most specific fractures encountered in non-accidental trauma (NAT) (Figure 11A).29-31 These occur due to anteroposterior compression of the chest wall, which levers the rib over the transverse process.30,32 This contrasts with lateral rib fractures, which are more common in non-abusive, accidental injuries.33,34 Oblique views of the ribs are crucial for detecting posterior rib fractures.
Other, less common, but highly specific chest wall fractures in NAT include acromial fractures, with a prevalence of roughly 5% among abused children (Figure 11B).28,35,36 Additional concerning injuries include non-acromial scapular fractures and non-birth related clavicular fractures. Recognizing these fractures on chest radiography is crucial, as these are often clinically occult, and identifying these lesions may permit appropriate management of a vulnerable child.
References
- Chowdhury MM, Chakraborty S. Imaging of congenital lung malformations. Semin Pediatr Surg. 2015;24(4):168-175. Doi: 10.1053/j.sempedsurg.2015.02.001.
- Laberge JM, Flageole H, Pugash D, et al. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: a Canadian experience. Fetal Diagn Ther. 2001;16(3):178-186.
- Wilson RD, Hedrick HL, Liechty KW, et al. Cystic adenomatoid malformation of the lung: review of genetics, prenatal diagnosis, and in utero treatment. Am J Med Genet A. 2006;140(2):151-155.
- Stocker Jt, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977; 8(2):155-171.
- Azizkhan RG, Crombleholme TM. Congenital cystic lung disease: contemporary antenatal and postnatal management. Pediatr Surg Int. 2008; 24(6):643-657.
- Nuchtern JG, Harberg FJ. Congenital lung cysts. Semin Pediatr Surg. 1994;3(4):233-243.
- Ozcan C, Celik A, Ural Z, et al. Primary pulmonary rhabdomyosarcoma arising within cystic adenomatoid malformation: a case report and review of the literature. J Pediatr Surg. 2001;36(7):1062-1065.
- Casagrande A, Pederiva F. Association between congenital lung malformations and lung tumors in children and adults: A systematic review. J Thorac Oncol. 2016;11(11):1837-1845.
- Ramos SG, Barbosa GH, Tavora FR, et al. Bronchioloalveolar carcinoma arising in a congenital pulmonary airway malformation in a child: case report with an update of this association. J Pediatr Surg. 2007;42(5): E1-4.
- d’Agostino S, Bonoldi E, Dante S, et al. Embryonal rhabdomyosarcoma of the lung arising in cystic adenomatoid malformation: case report and review of the literature. J Pediatr Surg. 1997;32(9):1381-1383.
- Koskas M, Balquet P, Tournier G, et al. Intralobar pulmonary sequestration manifested by neonatal cardiac insufficiency. Rev Mal Respir. 1991; 8(2):242-245.
- Pardes JG, Auh YH, Blomquist K, et al. CT diagnosis of congenital lobar emphysema. J Comput Assist Tomogr. 1983;7(6):1095-1097.
- Dillman JR, Sanchez R, Ladino-Torres MF, et al. Expanding upon the unilateral hyperlucent hemithorax in children. RadioGraphics. 2011; 31(3):723-741.
- Bagrodia N, Cassel S, Liao J, et al. Segmental resection for the treatment of congenital pulmonary malformations. J Pediatr Surg. 2014;49(6):905-909.
- Alamo L, Vial Y, Gengler C, et al. Imaging findings of bronchial atresia in fetuses, neonates and infants. Pediatr Radiol. 2016;46(3):383-390.
- Ranganath SH, Lee EY, Restrepo R, et al. Mediastinal masses in children. AJR Am J Roentgenol. 2012;198(3):W197-216.
- Ramos-Duran L, Nance JW, Schoepf UJ, et al. Developmental aortic arch anomalies in infants and children assessed with CT angiography. AJR Am J Roentgenol. 2012;198(5):W466-474.
- Billmire DF. Malignant germ cell tumors in childhood. Semin Pediatr Surg. 2006;15(1):30-36.
- Billmire D, Vinocur C, Rescorla F, et al. Malignant mediastinal germ cell tumors: an intergroup study. J Pediatr Surg. 2001;36(1):18-24.
- Horton Z, Schlatter M, Schultz S. Pediatric germ cell tumors. Surg Oncol. 2007;16(3):205-213.
- Rose RW, Ward BH. Spherical pneumonias in children simulating pulmonary and mediastinal masses. Radiology. 1973;106(1):179-182.
- Kim YW, Donnelly LF. Round pneumonia: imaging findings in a large series of children. Pediatr Radiol. 2007;37(12):1235-1240.
- Qureshi SS, Kembhavi S, Vora T, et al. Prognostic factors in primary nonmetastatic Ewing sarcoma of the rib in children and young adults. J Pediatr Surg. 2013;48(4):764-770.
- Murphey MD, Senchak LT, Mambalam PK, et al. From the radiologic pathology archives: Ewing sarcoma family of tumors: radiologic-pathologic correlation. Radiographics. 2013;33(3):803-831.
- Worch J, Matthay KK, Neuhaus J, et al. Ethnic and racial differences in patients with Ewing sarcoma. Cancer. 2010;116(4):983-988.
- Aurias A, Rimbaut C, Buffe D, et al. Translocation involving chromosome 22 in Ewing’s sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet Cytogenet. 1984;12(1):21-25.
- Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010;11(2):184-192.
- Barber I, Perez-Rossello JM, Wilson CR, et al. The yield of high-detail radiographic skeletal surveys in suspected infant abuse. Pediatr Radiol. 2015;45(1):69-80.
- Leventhal JM, Martin KD, Asnes AG, et al. Incidence of fractures attributable to abuse in young hospitalized children: results from analysis of a United States database. Pediatrics. 2008;122(3):599-604.
- Kleinman PK, Schlesinger AE. Mechanical factors associated with posterior rib fractures: laboratory and case studies. Pediatr Radiol. 1997;27(1):87-91.
- Maguire S. Which injuries may indicate child abuse? Arch Dis Child Educ Pract Ed. 2010;95(6): 170-177.
- Kleinman PK, Perez-Rossello JM, Newton AW, et al. Prevalence of the classic metaphyseal lesion in infants at low versus high risk for abuse. AJR Am J Roentgenol. 2011;197(4):1005-1008.
- Barsness KA, Cha ES, Bensard DD, et al. The positive predictive value of rib fractures as an indicator of nonaccidental trauma in children. J Trauma.2003;54(6):1107-1110.
- Bulloch B, Schubert CJ, Brophy PD, et al. Cause and clinical characteristics of rib fractures in infants. Pediatrics. 2000;105(4):E48.
- Lindberg DM, Harper NS, Laskey AL, et al. Prevalence of abusive fractures of the hands, feet, spine, or pelvis on skeletal survey: perhaps “uncommon” is more common than suggested. Pediatr Emerg Care. 2013;29(1):26-29.
- Kleinman PK, Morris NB, Makris J, et al. Yield of radiographic skeletal surveys for detection of hand, foot, and spine fractures in suspected child abuse. AJR Am J Roentgenol. 2013;200(3):641-644.
Related Articles
Citation
S S, J R, A H. Pediatric chest: A review of the must-know diagnoses. Appl Radiol. 2018; (4):4-14.
April 5, 2018