Juvenile granulosa cell tumor of the ovary

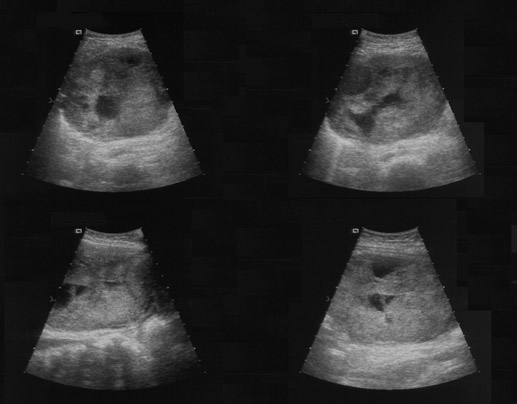
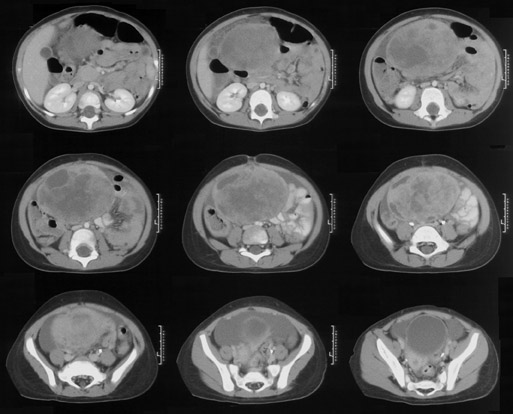
IMAGING FINDINGS
An abdominal radiograph was initially obtained and raised suspicion for a possible intussusception (Figure 1). A subsequent abdominalultrasound found a large complex low anteriorabdominal mass abutting the uterine fundus measuring 10 × 6.6 × 9.3 cm (Figure 2). Laboratory analysis revealed a dramatically elevated serum estradiol level.
Axial computed tomography (CT) of the abdomen and pelvis was performed following the administration of oral and intravenous contrast material. The enhancing mass demonstrated both solid and cystic components. There was free fluid within the abdomen and pelvis, but no lymphadenopathy. Prominent subareolar breast tissue was noted on images through the lower chest.
Based upon these findings, the patient underwent laparotomy for tumor resection and unilateral salpingo-oophorectomy.
DIAGNOSIS
Juvenile granulosa cell tumor of the ovary
DISCUSSION
Ovarian cancer is the third most common neoplasm of the female genital tract, after carcinoma of the cervix and endometrium. It accounts for 6% of all cancers diagnosed in females, and stands as the leading cause of gynecological cancer death. This is primarily because 80% of women with ovarian cancer present with advanced-stage disease.1
Based upon their cell type of origin, primary ovarian malignancies are classified into surface epithelium, germ cell, or sex cord tumors.Sex cord tumors account for 1% to 2% of ovarian malignancies. They may contain granulosa cells, thecal cells, Sertoli cells, or fibroblasts of gonadal stromal origin. While approximately two thirds occur in postmenopausal women, they can occur at any age, with 5% occurring in prepubescent patients. Approximately 70% of sex cord tumors are granulosa cell tumors. These tumors are often hormonally active. Thecal cells (luteinized cells within the stroma) produce estrogen, and if present in large enough quantities within a granulosa cell tumor, can cause elevated serum estradiol levels. Additionally, these tumors may also secrete inhibin. Ovarian granulosa cell tumors may also express unique cell surface markers that can be detected by staining sample tissue with histological markers. Surface expression of inhibin appears to have the greatest pathologic diagnostic potential for this tumor type.2
Among the histologic subtypes of granulosa cell tumors, juvenile granulosa cell tumors are rare, and typically present early. In a study of 125 patients with juvenile granulosa cell tumors, 78% presented within the first 2 decades of life.3Clinically, these patients typically presentwith signs of hyperestrogenism—precocious puberty in pre- pubertal patients, menstrual irregularities in women of reproductive age, and abnormal uterine bleeding in postmenopausal women.3 In addition, patients may complain of abdominal pain or an abdominal mass.
Management of granulosa cell tumors (including the juvenile subtype) begins with surgery for definitive tissue diagnosis, staging, and debulking. Surgical tumor removal typically includes ipsilateral salpingo-oophorectomy for patients wishing to retain fertility, and total hysterectomy for older patients or patients who no longer desire fertility. The role of chemotherapy or radiation therapy in the treatment of granulosa cell tumors remains uncertain because of the lack of prospective randomized trials supporting their roles as adjuvant agents.4 The paucity of extensive clinical trials reflects the overall rarity of these tumors. Prognosis depends upon surgical stage at presentation. Fortunately, most patients are diagnosed at stage I, and enjoy a favorable prognosis. Patients diagnosed at stages II through IV tend to have poor clinical outcomes with a higher rate of disease recurrence. Granulosa cell tumors in premenarchal girls appear to have a better outcome than those occuring in adult women.5
CONCLUSION
Juvenile granulosa cell tumors of the ovary represent a small fraction of all primary ovarian malignancies. They typically present within the first 2 decades of life with signs of hyperestrogenism, and an abdominal mass. As the majority of these tumors are diagnosed atstage I, their treatment remains surgical. The role of chemotherapy and/or radiation therapy remains unclear. Given the possibility for recurrence, long-term follow-up and surveillance is recommended for all patients.REFERENCES
- Funt SA, Hann LE. Detection and characterization of adnexal masses. Radiol Clin North Am. 2002;40:591-608.
- McCluggage WG. Recent advances in immunohistochemistry in the diagnosis of ovarian neoplasms. J Clin Pathol. 2000;53:327-334.
- Young RH, Dickersin GR, Scully RE. Juvenile granulosa cell tumor of the ovary. A clinicopathological analysis of 125 cases. Am J Surg Pathol. 1984;8:575-596.
- Schumer ST, Cannistra SA. Granulosa cell tumor of the ovary. J Clin Oncol. 2003;21:1180-1189.
- Lack EE, Perez-Atayde AR, Murthy AS, et al. Granulosa theca cell tumors in premenarchal girls: A clinical and pathologic study of ten cases. Cancer. 1981;48:1846-1854.