Congenital cystic lesions of the neck
Images














Dr. Mossa-Basha and Dr. Yousem are from the Russell H. Morgan Department of Radiology, Division of Neuroradiology, Johns Hopkins University, Baltimore, MD.
Cystic lesions of the head and neck span congenital, developmental, inflammatory, and vascular lesions. While these lesions may appear similar on computed tomography (CT) and magnetic resonance imaging (MRI) scans, their characteristic locations—midline or laterally, suprahyoid or infrahyoid, deep or superficial—may suggest a specific diagnosis. Other factors to consider include the lesions’ fluid density/intensity; whether they are unilocular or multilocular; whether they are deep or superficial to the carotid sheath; and whether they are associated with neck organs or with a specific patient population or syndrome.
This treatise will take a superior to inferior approach, beginning with skull base/intracranial lesions (arachnoid cysts), and concluding with mediastinal (thymopharyngeal), and lateral axillary lesions (lymphatic vascular malformations).
Arachnoid cysts
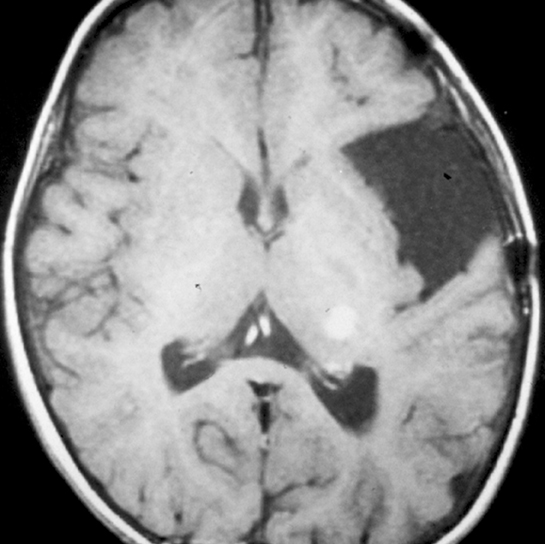
Arachnoid cysts are the most common extra-axial intracranial cystic lesions, representing 1% of all intracranial, space-occupying lesions. The true incidence of these lesions is unknown, however, as many of them are asymptomatic.1-5 Arachnoid cysts consist of a cerebrospinal fluid (CSF) collection enveloped by the arachnoid mater in the subarachnoid space.6 Intracranial arachnoid cysts are most commonly found in the middle cranial fossa (50% to 60%), the parasellar region (10% to 15%), the quadrigeminal plate cisterns (10%), the cerebellopontine angle (5% to 10%), the supracerebellar cistern (5%), and the cisterna magna (5%).7 Bilateral sylvian fissure arachnoid cysts should raise clinical suspicion for glutaric aciduria type 1, while posterior fossa arachnoid cysts are associated with Down syndrome.8,9 Arachnoid cysts can also be associated with mucopolysaccharidosis, neurofibromatosis type 1, schizencephaly, Aicardi syndrome, and Landau Kleffner syndrome.8,10 Syndromes associated with each congenital cystic lesion are listed in Table 1.
On CT imaging, arachnoid cysts typically appear as unilocular hypoattenuating lesions (similar to CSF in attenuation) in the extra-axial space, which can efface the underlying sulci and distort adjacent brain structures due to mass effect (Figure 1). The lesions can scallop the overlying bone. On conventional MRI sequences, arachnoid cysts typically follow CSF signal intensity on all sequences. The differential diagnosis includes epidermoid cysts, which generally can be distinguished by their bright signal, indicating reduced apparent diffusion coefficient on diffusion-weighted images. There can be underdevelopment of the underlying brain structures secondary to mass effect, especially those arachnoid cysts affecting the temporal lobe in the middle cranial fossa.8 Over time, arachnoid cysts can grow, remain stable, or involute.
Rathke’s cleft cysts
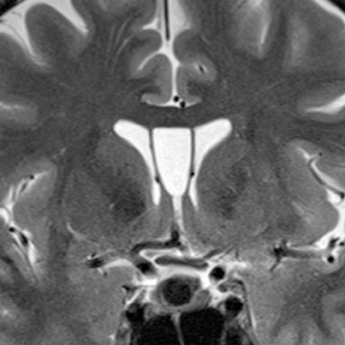
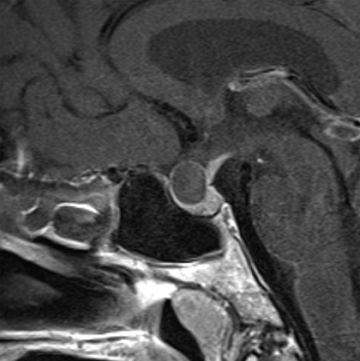
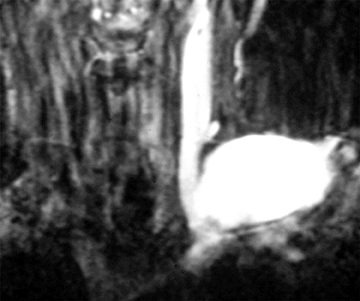
Rathke’s cleft cysts are congenital cysts that represent remnants of Rathke’s pouch, a dorsal invagination of the stomodeal neuroectoderm that ascends from the oral cavity into the sella to form the adenohypophysis. These non-neoplastic cysts are found on 11% to 22% of routine autopsies and are usually asymptomatic.11-13 While most Rathke’s cleft cysts remain asymptomatic throughout the patient’s life, occasionally patients can present due to visual symptoms, pituitary dwarfism, endocrine disturbances, or headaches.14-17
On noncontrast CT imaging, the cysts can be hyperdense, isodense, or hypodense.18 They are rarely suprasellar, but are more commonly intrasellar or both intrasellar and suprasellar.19 Intrasellar lesions are oval shaped, while intrasellar and suprasellar lesions can be shaped like dumbbells, with a waist at the diaphragm sella. The most common location of congenital cystic lesions of the head and neck is listed in Table 2. On MRI, the lesions can vary in T1 and T2 signal intensities, depending on their proteinaceous content, pure fluid, and/or blood products (Figure 2). The cysts should have thin, smooth walls without enhancing mural nodules. Thin cyst-wall enhancement can be seen (Figure 2), corresponding to chronic inflammatory changes thought related to foreign body reaction to cyst leakage of mucinous material. Rathke’s cleft cysts can have areas of enhancement along their margins, representing displaced and compressed pituitary parenchyma. T1-hyperintense and T2-hypointense nonenhancing intracystic nodules are seen in 11% to 77% of cases (Figure 2D).11,19 A phenomenon known as paradoxical enhancement has been described whereby the postcontrast cyst intensity may appear lower than the precontrast intensity.20 The typical imaging appearance of cystic lesions of the neck is described in Table 3.
Occipital encephaloceles
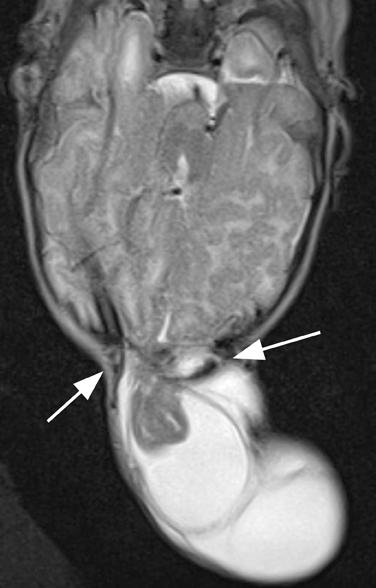
A cephalocele/encephalocele represents a malformation in which intracranial contents (CSF, meninges, and brain parenchyma) protrude through a cranial defect. Neurodevelopment in the setting of a cephalocele, in which brain tissue herniates through the defect, depends on the size of the lesion, the amount of herniated cerebral tissue, and associated anomalies. The incidence of encephaloceles is estimated to be 1 to 4 per 10,000.21-23 The most common types within the United States (U.S.) and European populations are occipital encephaloceles; they represent 75% of all encephaloceles amongst the U.S. population, and are commonly associated with conditions, such as hemifacial microsomia; Joubert, Chemke, Knobloch, and von Voss syndromes; and Chiari III, Meckel Gruber and Dandy-Walker malformations.7,24,25 Occipital encephaloceles are frequently associated with abnormalities of the dural venous sinuses, including absence of the straight sinus and a persistent falcine sinus.26,27 These lesions can be associated with hydrocephalus or aventriculy.7,24 Occipital encephalocele is typically clinically evident (sincipital). Imaging studies are ordered to determine whether there are associated anomalies, and if the dural venous sinuses course into the encephalocele sac (Figure 3) to avoid surgical resection.
Sincipital encephaloceles, nasal gliomas, dermal sinus tract, and nasal dermoids
Sincipital (frontonasal, nasoorbital, and nasoethmoidal) encephaloceles, nasal dermoids, nasal gliomas, and frontoethmoidal sinus tracts all represent a spectrum of congenital anomalies. They all arise from a lack of regression of a dural diverticulum in the prenasal space that extends through the embryologic foramen cecum or the fonticulus frontalis.28 If this dural projection extends to the skin surface, dermal elements will be pulled in as the diverticulum retracts intracranially, forming a dermal sinus tract. Dermoid or epidermoid cysts can develop anywhere along this sinus tract, from the nasal soft tissues to the foramen cecum adjacent to the crista galli. Sincipital encephaloceles result from herniation of intracranial contents through the dural projection and into the nasal cavity. Nasal gliomas (a misnomer as they are not actually tumors) represent dysplastic heterotopic brain tissue in the nasal cavity or extranasally in the subcutaneous tissues, which have embryologically herniated through the dural cleft that later regressed.7
Sincipital encephaloceles are the most common type of encephalocele in Southeast Asia, and their occurrence in the U.S. has rapidly increased due to Asian immigration.29 Frontal encephaloceles, which include sincipital (outwardly apparent) and basal (not visible externally) encephaloceles are the second-most common encephalocele, representing 25% of cases.28 Clinical presentation can include CSF leak, rhinorrhea, and meningitis.28,30 Frontal encephaloceles can be associated with frontonasal dysplasia, aberrant tissue band syndrome, and Morning Glory syndrome.25,31
Critical to the evaluation of congenital nasal masses is whether there is communication or involvement of the intracranial structures.32 MRI is typically the modality of choice, evaluating the lesion as well as looking for additional intracranial anomalies. On MRI, sincipital encephalocele will present as a glabellar, prenasal, nasal, or nasal septum mass, which is continuous with the intracranial contents, contains CSF signal intensity fluid and brain parenchyma (which may be T2 hyperintense secondary to gliosis). There can be associated osseous abnormalities, including widening of the foramen cecum, absence of the cribiform plate or crista galli with frontoethmoidal encephalocele, or a defect in the fonticulus frontalis in the setting of frontonasal encephalocele (figure 4). Nasal gliomas can occur anywhere along the primitive dural projection, from the nasal soft tissues to the foramen cecum, similar to dermoid/epidermoid. On MRI, nasal gliomas are nonenhancing lesions within the nose or anterior to the nasofrontal suture (actually extranasal) or nasal septum that are T1 isointense and slightly T2 hyperintense (secondary to gliosis), relative to brain parenchyma. On MRI, nasal dermal sinus tracts will present as T1-hypointense linear tract extending from the foramen cecum through the subcutaneous soft tissues of the midline nasal bridge to the prenasal space. As stated previously, dermoids or epidermoids can occur anywhere along this tract, and can even occur intracranially. Epidermoids will typically follow fluid signal on T1- and T2-weighted sequences, but will show diffusion restriction, similar to epidermoids elsewhere in the body. Dermoids will contain areas of T1 shortening and variable T2 signal secondary to intralesional fat.
Sebaceous and epidermal cysts
The sebaceous or epidermal inclusion cyst is a common finding in the skin and subcutaneous tissue in asymptomatic patients. The lesions may be palpable to the patient and are usually unilocular and ovoid in shape.33 Lipomas may be in the differential diagnosis. On imaging these lesions, they have cystic density but also variable T1W signal intensity based on their protein/lipid/keratin content. They have rim enhancement and are generally bright to mixed intensity on T2W images. Rupture of these lesions into the skin may induce a local cellulitis, but rarely evolves into abscesses. Ruptured cysts have a higher rate of septation, thick and irregular rim enhancement, and spread of abnormal signal and enhancement into surrounding subcutaneous tissues.
Thornwaldt cysts
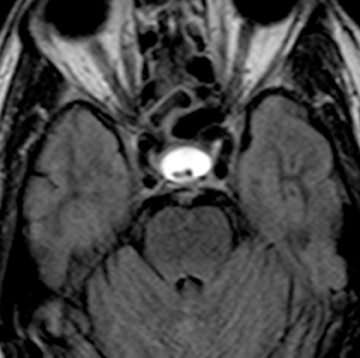
Thornwaldt cysts represent benign congenital lesions that arise in the midline of the nasopharynx, seen in approximately 0.2% to 5% of the population on MRI studies.34,35 These lesions arise at the contact point between the embryonic notochord and the nasopharyngeal ectoderm.34 There is retraction of nasopharyngeal mucosa with the notocordal ascent into the clivus. Thornwaldt cysts are typically unilocular and hypoattenuating on CT. On MRI, they are T2 hyperintense and of variable T1 signal intensity, depending on proteinaceous content (Figure 5). There can be thin peripheral cyst-wall enhancement on postcontrast imaging. Thornwaldt cysts are typically asymptomatic, but they can result in occipital headaches, persistent nasal discharge, or halitosis in the setting of infection.36
First branchial cleft cysts
The fetal branchial apparatus develops in the 2nd gestational week and consists of 6 pairs of mesodermal arches separated by 5 ectoderm-lined branchial clefts externally and 5 endoderm-lined pharyngeal pouches internally.37 By the 4th week of embryonic life, 4-paired mesodermal arches are apparent externally, with smaller 5th and 6th branchial arches not well visualized.38 The 2nd branchial arch enlarges caudally, extending toward the enlarging epicardial ridge of the 5th branchial arch. This leads to enclosure of the 2nd, 3rd, and 4th branchial clefts in an ectoderm-lined cavity known as the cervical sinus of His, which ultimately involutes.39 Branchial anomalies represent 30% of all congenital neck lesions.40 The most commonly discussed branchial apparatus anomalies are cysts, fistulas and sinus tracts, but they also include thymic cysts, aberrant parathyroid tissue, and parathyroid cysts.38 The most commonly accepted theory of anomalous development is persistence of trapped cell rests from incompletely obliterated branchial apparatus, resulting in development of branchial cleft cysts later in life. On the other hand, persistence of a branchial cleft, pouch, or the sinus of His can produce a branchial sinus or fistula.41 A branchial sinus represents a tract that communicates externally, and only rarely internally. A branchial fistula communicates both internally (pharyngeal mucosa) and externally (neck surface), and relates to persistence of both a branchial cleft and pouch, while branchial cysts do not communicate with external or internal surfaces.39 The most common branchial cleft anomalies are cysts, representing 75% of all cases, while 25% are fistulas and sinuses.42
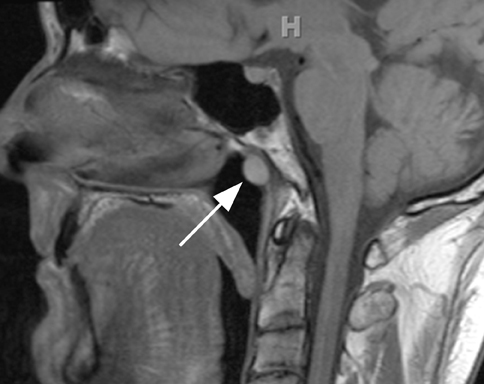
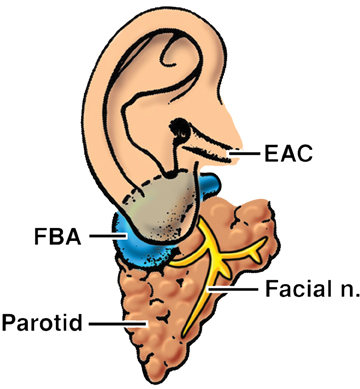
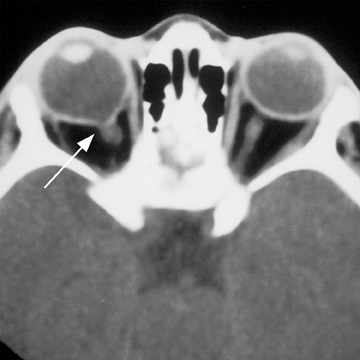
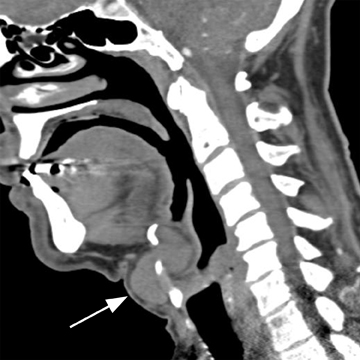
The first branchial apparatus normally gives rise to the eustachian tube, tympanic cavity, mastoid antrum, a portion of the tympanic membrane, external auditory canal, the mandible, maxilla, and the malleus and incus.43 For this reason, first branchial cleft cysts, which represent the second-most common branchial cleft anomaly, can arise anywhere from the external auditory canal through the parotid gland to the submandibular triangle. They are associated with branchio-oto-renal syndrome.44 First branchial cleft cysts most commonly present with recurrent or refractory abscesses in the periauricular or parotid region, or at the angle of the mandible. Cysts that communicate with the external auditory canal can present with otitis, otorrhea and/or auricular swelling.43 Intraparotid lesions can also present with parotitis. Complete surgical excision is curative. Arnot type I cysts are located within the parotid gland without a fistula to the external auditory canal (Figure 6). Arnot type II cysts are typically inferior to the parotid gland and may have a fistulous connection to the ear (Figure 6).
First branchial cleft cysts appear as simple or complicated unilocular cystic lesions within, superficial, or deep to the parotid gland on CT and MRI (Figure 7). Cyst-wall thickness and enhancement varies, depending on whether the lesion is infected. The imaging appearance is nonspecific and differential considerations include cystic parotid neoplasms, lymphadenopathy, sialocele, pseudocyst, retention cyst, or AIDS-/Sjogren’s-related lymphoepithelial cysts.41
Dacrocystoceles
Nasolacrimal duct obstruction (NLDO) is the most common lacrimal abnormality in infants and children, occurring in 2% to 30% of newborns, most commonly secondary to incomplete canalization of the distal nasolacrimal duct, specifically the valve of Hasner.39,45,46 Dacrocystoceles, which occur in 0.1% of NLDO cases, represent mucinous distention of the lacrimal sac, with mucinous material secondary to congenital obstruction of the nasolacrimal apparatus both proximally and distally.47 There may be marked dilatation of the nasolacrimal duct and lacrimal sac, which appears as a CSF-intensity lesion on CT and MRI. There can be thin wall enhancement. When infected, dacrocystocele can appear as a complicated cyst with preseptal soft-tissue swelling.
Dacrocystoceles are usually benign, but if bilateral, they can rarely result in stenosis of the nasal aperture, leading to respiratory distress.45 Patients most commonly present shortly after birth with a bluish, subcutaneous cystic swelling inferior to the medial canthus or a nasal mass.48
Staphylomas
Staphylomas represent congenital or acquired areas of scleral insufficiency, secondary to degenerative, traumatic or inflammatory injury, resulting in focal outward bulging of the globe. Staphylomas are usually lined by choroidal tissue. These lesions may be isolated findings, or they can be associated with retinal detachment or severe axial myopia.39 Staphylomas can occur anywhere on the globe, with a focal globe bulge in a region of uveoscleral thinning. Staphyloma can be associated with epidermal nevus syndrome and MRCS (microcornea, rod-cone dystrophy, cataract, and posterior staphyloma).49,50
Colobomas
Ocular colobomas are developmental abnormalities resulting from incomplete closure of the embryonic choroidal fissure. They may affect any part of the globe, but they favor the optic nerve insertion site.51 These lesions are bilateral in 60% of cases and the globes can be normal-sized or small.52 The most common type involves the inferior temporal aspect of the iris and is not readily seen on imaging.29 There can be involvement of the optic nerve, retina, choroid, iris, or lens, and when all of these structures are involved, it is known as a complete coloboma. Coloboma can involve the optic nerve at the optic disk (Figure 8), with resultant enlargement and excavation of the optic disk. Microophthalmia with cyst represents a severe malformation in which a prominent retrobulbar cyst may communicate with the vitreous cavity, allowing for herniation of vitreous humor through the defect into the cyst.53 The relative size of the globe and cyst are variable in this setting.
Colobomas can occur as an isolated congenital abnormality or as part of a syndromic setting, including CHARGE (Coloboma, Heart malformations, choanal AtResia, Growth and/or mental retardation, genital anomalies, Ear anomalies and/or deafness), VATER (Vertebrae, Anus, Trachea, Esophagus, and Renal), branchio-oculo facial, Meckel-Gruber, Walker-Warburg, and Goldenhar syndromes; Aicardi, basal cell nevus; and Trisomy 13 and 18.54,55
Thyroglossal duct cysts
Thyroglossal duct cysts (TGDC) represent 70% of all congenital neck masses and are the most common congenital cervical abnormality after Thornwaldt cysts.56 During the 4th gestational week, the thyroid primordia, which originates as a diverticulum in the floor of the primitive pharynx, grows caudally from the foramen cecum at the junction of the oral tongue and the base of the tongue, through the prepharyngeal soft tissues along a midline descent, the anterior neck, anterior and in close proximity to the developing hyoid bone, to its eventual destination in the inferior neck.40 As the developing gland travels caudally, an epithelial tract is left behind that develops into the thyroglossal duct, extending from the foramen cecum, anterior to the hyoid bone, thyrohyoid membrane and possibly embedded in the strap muscles and to the inferior neck in the midline.41 The thyroglossal duct frequently forms a recurrent loop behind the hyoid bone.57 For this reason, TGDC can extend through or around the hyoid bone (Figure 9). The duct generally involutes by the 5th to 10th week of gestation, leaving only a proximal remnant at the foramen cecum, and a distal portion that differentiates into the pyramidal lobe of the thyroid gland.40 Persistence of any portion of the duct can lead to the formation of cysts or ectopic thyroid rests along the tract. The Sistrunk procedure is performed for removal of TGDC, in which the cyst, the entire thyroglossal tract, and the central hyoid bone are removed.
On all imaging modalities, TGDCs appear as simple unilocular cystic lesions that are most commonly located juxtahyoid or infrahyoid.58,59 Seventy-five percent of TGDCs occur in the midline, while 25% are within 2 cm of midline in the anterior neck.58 Fifteen percent occur at the level of the hyoid bone, while 60% occur just inferior to the hyoid, embedded within the strap muscles.43 Thyroglossal duct cysts can have a thin, peripheral, enhancing cyst wall after intravenous contrast administration. On MRI, intrinsic T1 and T2 signal varies depending on the cyst protein/colloid content, but is usually bright on both.60 Complicated cysts with increased attenuation, internal septations, and a thick rim of enhancement can result after prior infection or hemorrhage. A solid enhancing nodule or calcifications within the cyst should raise suspicion for functioning tissue (1% to 2% of cases) and/or thyroid carcinoma (most commonly papillary), which arises in < 1% of all lesions (Figure 10).61 Thyroglossal duct cysts as well as other thyroid anomalies occur in the setting of Cowden syndrome.62
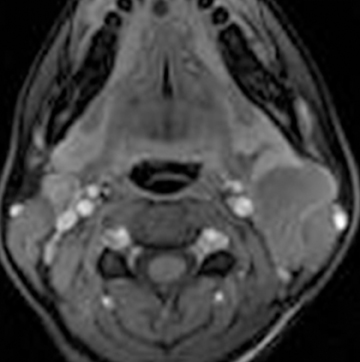
Second branchial cleft cysts
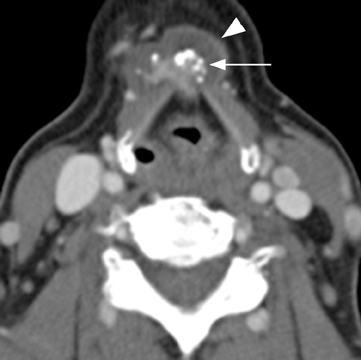
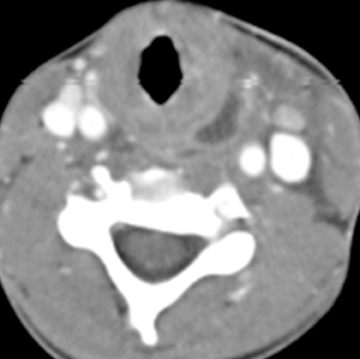
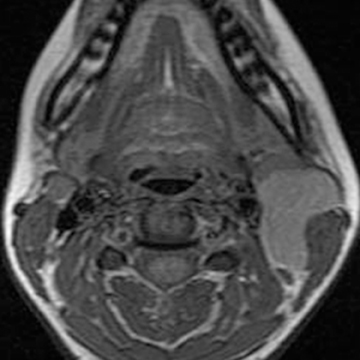
Second branchial cleft anomalies are the most common branchial apparatus deformities, representing 95% of all lesions, with cysts being more common than fistulae and sinuses.63 These lesions are thought to arise from incomplete obliteration of the embryologic cervical sinus of His. Second branchial cleft cysts are most commonly found at the angle of the mandible along the anteromedial border of the sternocleidomastoid muscle, but can occur anywhere from the oropharyngeal tonsillar fossa to the supraclavicular neck.64 Second branchial cleft cysts are classified into 4 Bailey types, based on location (Figure 11).65 Type 1 anomalies are anterior to the sternocleidomastoid muscle and deep to the platysma (Figure 11), while type 2 lesions, which are the most common type, are found deep to the sternocleidomastoid muscle, superficial to the carotid sheath (Figures 11 and 12). Type 3 lesions extend through the carotid bifurcation close to the lateral wall of the pharynx (Figures 11 and 12). Type 4 lesions are situated adjacent to the tonsillar fossa, medial to the carotid sheath, where they may present as parapharyngeal masses (Figure 11). On CT, these lesions are typically well-circumscribed, homogeneously hypoattenuating, unilocular cystic lesions, while on MRI, branchial cleft cysts can be T1 hypointense to intermediate intensity relative to muscle, and are typically T2 hyperintense, similar to CSF intensity, unless traumatized or infected. Mural thickness, the degree of cyst wall enhancement, complexity of the lesion, and adjacent soft-tissue edema depend on the extent of associated inflammation/infection. They are associated with branchio-oto-renal syndrome and branchio-oculo-facial syndrome.44,66 Metastatic squamous cell lymph nodes can have an appearance similar to second branchial cleft cysts and should be a consideration in the appropriate setting. There is a propensity for such cystic lymph nodes in cases of tonsillar carcinoma and/or HPV-related squamous cell carcinomas.
Dermoids/epidermoids of the floor of the mouth
Dermoids and epidermoids are the second-most common midline cervical anomalies after thyroglossal duct cysts.67 Dermoid and epidermoid lesions are separable by the presence of epithelial appendages within dermoid cyst walls. Epidermoid cysts are much less common in the head and neck, and present earlier, usually during infancy, compared to dermoid cysts.43 Epidermoid cysts of the head and neck can be associated with Gardner’s syndrome, with the lesions frequently affecting the scalp, face, and soft tissues of the neck.68 Dermoid cysts can occur anywhere within the body, with only 7% occurring in the head and neck, most commonly involving the lateral brow and nasal regions (as discussed above).43 Approximately 11.5% of dermoid cysts are located in the midline of the floor of the mouth or within the oral tongue.
On imaging studies, epidermoid cysts will present as thin-walled cystic lesions in the submandibular or sublingual space (Figure 13). On CT, these lesions typically have simple fluid attenuation, with rim enhancement after intravenous contrast administration. DWI studies show restricted diffusion, as in intracranial lesions. Alternatively, dermoids may have fat-fluid and fluid-fluid levels or the lesion may have a heterogeneous appearance due to the various germinal components. There may be the appearance of intracystic rounded lesions, relating to coalescence of fat nodules within the fluid matrix, giving the appearance of “a sack of marbles.”69 This finding is virtually pathognomonic for dermoid cysts. MR imaging permits evaluation of lesion extent in the tongue and floor of the mouth, and the lesion’s relationship to adjacent muscles. Most dermoid cysts are located superior to the mylohyoid muscle.40 Dermoid cysts are T1 hyper- to iso-intense, depending on sebaceous lipid content, and are generally T2 hyperintense.
Third branchial cleft cysts
Although third branchial cleft anomalies are rare, they are still the second most common congenital lesion of the posterior cervical triangle of the neck after lymphatic malformations.70 Most third branchial cleft cysts are diagnosed during childhood, and 97% are located on the left side.71 Third and fourth branchial cleft cysts commonly present with recurrent neck infections (Figure 14). Differentiating third and fourth branchial cleft cysts from each other can be difficult due to close proximity, but the relationship of the sinus tract to the superior laryngeal nerve (derived from the 4th branchial arch) surgically is helpful. Third branchial cleft anomalies lie superior to the superior laryngeal nerve, while fourth branchial anomalies lie below. Third branchial cleft cysts are typically located posterior to the common carotid or internal carotid artery and sternocleidomastoid (within the posterior cervical triangle), superior to the hypoglossal nerve, and inferior to the glossopharyngeal nerve.43 A complete third branchial fistula has external communication anterior to the sternocleidomastoid muscle. The tract courses posterior to the common or internal carotid artery, anterior to the vagus nerve, and pierces the thyrohyoid membrane.38 Traditionally, third branchial cleft anomalies are depicted entering the base of the pyriform sinus anterior to the fold of the internal laryngeal nerve; however, Rea et al have described cases in which the anomaly occasionally originates from the apex of the pyriform sinus.72
Third branchial cleft cysts most commonly appear on CT and MRI as unilocular cystic lesions in the posterior cervical triangle. The extent of cyst-wall thickness and enhancement, cyst complexity and adjacent edema depends on whether superimposed or previous inflammation/infection and/or fistulous tracts (which are often superinfected) are present. There is variable T1-signal intensity depending on the proteinaceous content, but the lesions are typically T2 hyperintense. Differential considerations include fourth branchial cleft cysts, cystic/necrotic lymph nodes, and lymphatic malformations. The definitive treatment is surgical excision.71
Fourth branchial cleft cysts
Fourth branchial cleft anomalies are very rare lesions, representing 1% to 4% of all branchial cleft anomalies.73,74 These lesions typically present during early childhood, much like third branchial cleft anomalies, typically secondary to superimposed, acute, suppurative thyroiditis, or recurrent abscess formation.75 Fourth branchial cleft anomalies are most commonly sinus tracts and are seen on the left.43 The sinus tract courses from the apex of the pyriform sinus medial and inferior to the recurrent laryngeal nerve, loops superior to the hypoglossal nerve, travels along the course of the recurrent laryngeal nerve within the trachesophageal groove, wraps around the aortic arch on the left or the subclavian artery on the right. The tract then courses superiorly dorsal to the common carotid artery, loops around the hypoglossal nerve and extends medially to the sternocleidomastoid muscle.76 This depends on the length of the 4th branchial cleft anomaly, as most do not complete the above mentioned course. Surgical excision of these lesions with partial thyroidectomy is the definitive treatment.75
Thymic and thymopharyngeal duct cysts
Thymic cysts are very rare cervical masses usually detected during childhood that can arise within the neck or anterior mediastinum, occurring along the course of thymic descent. Thymic cysts can occur anywhere from the angle of the mandible, along the lateral neck, to the sternal notch, but cervical thymic cysts occur more frequently in the lower third of the neck on the left side anterior to the sternocleidomastoid muscle. Overall, these lesions occur much more commonly in the mediastinum than in the neck (25:1 ratio).77 Nearly half of all cervical thymic cysts are continuous with the mediastinal thymus, either via mediastinal extension of the cyst or by a vestigial remnant of thymic tissue or a solid cord.78 Embryologically, the thymus originates from the 3rd and 4th pharyngeal pouches. During the 6th week, as the paired thymic buds develop from ventral outgrowths, the thymopharyngeal duct forms caudally along the course of migration of the thymus into the mediastinum.79,80 During the 7th to 10th week, as the embryologic thyroid migrates into the mediastinum, the proximal thymopharyngeal duct atrophies and separates from the pharynx.79 Residual thymic tissue or thymic anomalies can arise anywhere along the thymopharyngeal duct. There are multiple theories regarding the development of thymic cysts, but 2 of the more commonly held theories will be discussed here.79 The first believes that thymic cysts arise from persistence of portions of the thymopharyngeal duct. The second theory is based upon cystic degeneration of Hassall’s corpuscles within ectopic thymic tissue within the neck or anterior mediastinum. Thymic cysts and thymopharyngeal duct cysts are sometimes used interchangeably, but for our purposes we will use these terms to refer to a spectrum of disease, with thymopharyngeal duct cyst referring to a lesion spanning the entire course of the embryologic tract. These lesions most often present as painless, slowly growing masses in the lateral neck at the thoracic inlet, close to the thoracic duct insertion to the subclavian artery.77 Patients can, however, present with symptoms including upper respiratory infection, coughing, hoarseness, dysphagia, wheezing, and even respiratory distress secondary to tracheal compression.81 Thymic cysts usually present between the ages of 2 and 13 years, later than lymphangioma and earlier than branchial cleft cysts.79
Preoperative diagnosis of thymic cysts is difficult, as these lesions are rare and frequently confused for branchial cleft cysts or lymphangiomas. Cervical thymic cysts most commonly occur in the lower neck, compared to branchial cleft cysts that are most often seen in the upper neck.81 Second branchial cleft cysts usually pass between the external and internal carotid arteries, while cervical thymic anomalies will pass between the common carotid artery and the vagus nerve, based on the embryologic development of the 3rd pharyngeal pouch.78 There is mediastinal extension in half of cervical thymic cysts while in contradistinction, only 3% to 10% of lymphangiomas extend into the mediastinum.81 Lymphangiomas most commonly occur in the posterior cervical triangle. Thymic cysts are usually unilocular (or less likely multilocular) elongated cysts, frequently tapered at their ends, and following fluid signal on CT and MR imaging (Figure 15).78 There can be a thin rim of cyst-wall enhancement on postcontrast imaging. Complete surgical excision, even with asymptomatic lesions, is used for diagnosis and treatment. Confirmation of the presence of mediastinal thymic tissue should be performed prior to removal of thymic cysts or thymic ectopic tissue to prevent immunocompromise.79 The diagnosis is established by histologic demonstration of residual thymic tissue and Hassall’s corpuscles in the cyst wall. Congenital thymic cysts should be differentiated from multilocular thymic cyst, as the latter represents an acquired, multilocular, inflammatory lesion arising from cystic dilatation of the medullary duct and having an association with autoimmune diseases, such as Sjogren’s syndrome and aplastic anemia.82
Lymphatic vascular malformations
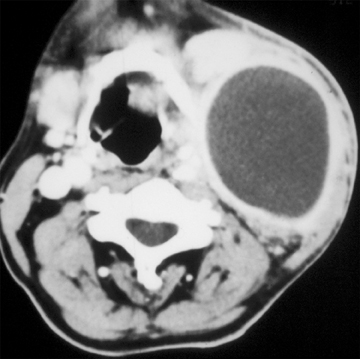
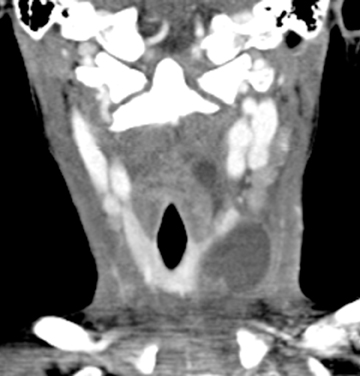
Lymphatic vascular malformations are developmental anomalies of the lymphatic system that can occur anywhere in the body, but 75% affect the head and neck.40 Lymphangiomas of the head and neck most commonly occur in the posterior cervical triangle followed by the axilla and the tongue musculature.83 Lymphangiomas are thought to develop from sequestration of a portion of the embryologic lymphatics (most commonly involving the jugular lymph sac), resulting in continued growth and accumulation of fluid.84 Alternatively, a lymphangioma may arise from failure of the juguloaxillary lymphatic sac to drain into the venous system, resulting in obstruction of the lymphatic channel. Lymphatic vascular malformations are categorized into 3 different histologic subtypes, determined by the size of the lymphatic channels. Simple or microcystic lymphangiomas have capillary-sized, thin-walled lymphatic channels, while cavernous lymphangiomas are composed of dilated lymphatic spaces and fibrous adventitia. The third type, cystic hygroma, is a multilocular cystic lesion composed of cysts of varying sizes.85 All 3 histologic types can occur within the same lesion. These malformations are commonly detected before the age of 2 years, presenting as slow-growing asymptomatic lesions, but sudden enlargement can occur in the setting of infection, intralesional hemorrhage, trauma, or even respiratory tract infection.43 Half are discovered at birth and can present with respiratory compromise or even death secondary to mass effect on the airway. Hybrid lesions with venous components are not unusual as low flow vascular malformations.
Lymphangiomas of the head and neck most commonly occur in the posterior cervical triangle, with 3% to 10% extending into the mediastinum, and 20% involving the axilla.40 The malformations may also occur in the submandibular space. Lymphangiomas are frequently transspatial due to their infiltrative nature and do not respect fascial planes. The size of these lesions is highly variable. The extent of the lesion is best evaluated on MRI. On MRI, these multiloculated cystic lesions can have low to intermediate T1 signal, but occasionally increased T1 signal (Figure 16) secondary to intralesional hemorrhage, high protein, or fat content. There can be rim enhancement as well (Figure 16). They uniformly have T2 hyperintensity unless they have acutely bled. Fluid-fluid levels may be present secondary to blood. On CT, a lymphangioma can appear as a poorly circumscribed, homogeneous fluid attenuation mass that may contain intralesional blood. Infected lesions may contain higher attenuation fluid. If they are combined with venous vascular malformations, they may have enhancing solid components. Lymphatic malformations are associated with fetal alcohol, Turner’s, Down, and Noonan’s syndromes.
Thoracic duct/jugular lymphatic cysts
The thoracic duct is the largest lymphatic vessel in the body, draining lymph and chyle from the subdiaphragmatic body, left arm and chest (except for the lower lobe of the left lung) into the left brachiocephalic vein. The thoracic duct arises from the cisterna chyli in the right abdomen, crosses the diaphragm, and courses cranially just right of the midline. The duct crosses over to the left at the level of the 4th vertebral body and passes into the neck, where it forms an arch just above the clavicle before draining into the left subclavian vein.86 Embryologically, the thoracic duct develops from 6 lymph sacs, with paired jugular lymph sacs arising at the junction of the internal jugular veins and subclavian veins.86 Thoracic duct cysts most commonly involve the cisterna chyli, followed by the mediastinum and rarely involve the neck.87 Cervical thoracic duct lymphoceles are rarely congenital, more commonly arising secondary to thoracic duct injury after neck dissection or trauma.88 The exact pathophysiologic development of thoracic duct cysts is incompletely understood. Congenital weakness of the duct wall or obstruction at the lymphatic-venous junction may contribute to their development.89 Lymphatic duct wall degeneration secondary to atherosclerosis, trauma, or infection may also play a developmental role. Persistence of the fetal jugular lymphatic sac is thought to result from incompetence or absence of the valve at the lymphatic-venous junction, thus allowing reflux of blood into the thoracic duct.88 Thoracic duct cysts typically present with a painless fluctuant left supraclavicular mass, but they can also present secondary to mass effect on adjacent structures.
Cervical thoracic duct cysts typically occur adjacent to the confluence of the left subclavian and internal jugular veins in the supraclavicular fossa, deep to the sternocleidomastoid muscle, and lateral to the carotid sheath. MRI is optimal for delineating the extent of the lesion and will show a unilocular or multilocular cystic lesion in the left supraclavicular fossa with possible retrosternal extension (Figure 17). Lymphangiography (the gold standard exam) or lymphoscintigraphy can be performed to confirm communication with the thoracic duct. The diagnosis is established by fine needle aspiration with return of milky fluid and biochemical fluid analysis. Asymptomatic lesions can be treated conservatively, but in the symptomatic setting surgical resection with thoracic duct ligation or cyst aspiration with sclerosant injection can be performed.90
Conclusion
Congenital cystic lesions of the neck are infrequently encountered disease processes, which span congenital, developmental, inflammatory, and vascular lesions. While there may be overlap in cross-sectional imaging appearances, characteristic locations, cyst fluid attenuation/signal intensity, association with adjacent structures, and cyst features can help in differentiating these various lesions. Imaging also allows for biopsy and surgical planning.
References
- Aicardi J, Bauman F. Supratentorial extracerebral cysts in infants and children. J Neurol Neurosurg Psychiatry. 1975;38:57-68.
- Leo JS, Pinto RS, Hulvat GF, et aI. Computed tomography of arachnoid cysts. Radiology. 1979;130:675-680.
- Parsch CS, Krauss J, Hofmann E, et al. Arachnoid cysts associated with subdural hematomas and hygromas: Analysis of 16 cases, long-term follow-up, and review of the literature. Neurosurgery. 1997;40:483-490.
- Passero S, Filosomi G, Cioni R, et al. Arachnoid cysts of the middle cranial fossa: A clinical, radiological and follow-up study. Acta Neurol Scand. 1990;82:94-100.
- von Wild K. Arachnoid cysts of the middle cranial fossa. Neurochirurgia (Stuttg). 1992;35: 177-182.
- Yildiz H, Erdogan C, Yalcin R, et al. Evaluation of communication between intracranial arachnoid cysts and cisterns with phase-contrast cine MR imaging. AJNR Am J Neuroradiol. 2005;26:145-151.
- Castillo M. Neuroradiology. Philadelphia: Lippincott Williams and Wilkins; 2002.
- Gosalakkal JA. Intracranial arachnoid cysts in children: A review of pathogenesis, clinical features, and management. Pediatr Neurol. 2002;26:93-98.
- Pearlson GD, Breiter SN, Aylward EH, et al. MRI brain changes in subjects with Down syndrome with and without dementia. Dev Med Child Neurol. 1998;40:326-334.
- Zubairi MS, Carter RF, Ronen GM. A male phenotype with Aicardi syndrome. J Child Neurol. 2009;24:204-207.
- Byun WM, Kim OL, Kim D. MR imaging findings of Rathke’s cleft cysts: Significance of intracystic nodules. AJNR Am J Neuroradiol. 2000;21:485-488.
- Isono M, Kamida T, Kobayashi H, et al. Clinical features of symptomatic Rathke’s cleft cyst. Clin Neurol Neurosurg. 2001;103:96-100.
- Tominaga JY, Higano S, Takahashi S. Characteristics of Rathke’s cleft cyst in MR imaging. Magn Reson Med Sci. 1 2003;2:1-8.
- Benveniste RJ, King WA, Walsh J, et al. Surgery for Rathke cleft cysts: Technical considerations and outcomes. J Neurosurg. 2004;101: 577-584.
- Eguchi K, Uozumi T, Arita K, et al. Pituitary function in patients with Rathke’s cleft cyst: Significance of surgical management. Endocr J. 1994;41:535-540.
- el-Mahdy W, Powell M. Transsphenoidal management of 28 symptomatic Rathke’s cleft cysts, with special reference to visual and hormonal recovery. Neurosurgery. 1998;42:7-16; discussion 16-17.
- Kim JE, Kim JH, Kim OL, et al. Surgical treatment of symptomatic Rathke cleft cysts: Clinical features and results with special attention to recurrence. J Neurosurg. 2004;100:33-40.
- Nishioka H, Haraoka J, Izawa H, Ikeda Y. Magnetic resonance imaging, clinical manifestations, and management of Rathke’s cleft cyst. Clin Endocrinol (Oxf). 2006;64:184-188.
- Wen L, Hu LB, Feng XY, et al. Rathke’s cleft cyst: Clinicopathological and MRI findings in 22 patients. Clin Radiol. 2010;65:47-55.
- Yousem DM, Ihmeidan I, Quencer R, Atlas SW. Paradoxically decreased signal intensity on postcontrast short-TR MR images. AJNR Am J Neuroradiol. 1991;12:875-880.
- Antunes JL, Sharer LR, Pellock JM. Occipital encephalocele—a case of conjoined twinning? Neurosurgery. 1983;13:703-707.
- Kotil K, Kilinc B, Bilge T. Diagnosis and management of large occipitocervical cephaloceles: A 10-year experience. Pediatr Neurosurg. 2008;44:193-198.
- Raja RA, Qureshi AA, Memon AR, et al. Pattern of encephaloceles: A case series. J Ayub Med Coll Abbottabad. 2008;20:125-128.
- Kiymaz N, Yilmaz N, Demir I, Keskin S. Prognostic factors in patients with occipital encephalocele. Pediatr Neurosurg. 2010;46:6-11.
- Cohen MM, Jr., Lemire RJ. Syndromes with cephaloceles. Teratology. 1982;25:161-172.
- Bartels RH, Merx JL, van Overbeeke JJ. Falcine sinus and occipital encephalocele: A magnetic resonance venography study. J Neurosurg. 1998;89:738-741.
- Otsubo Y, Sato H, Sato N, Ito H. Cephaloceles and abnormal venous drainage. Childs Nerv Syst. 1999;15:329-332.
- Rahbar R, Resto VA, Robson CD, et al. Nasal glioma and encephalocele: Diagnosis and management. Laryngoscope. 2003;113:2069-2077.
- Barkovich AJ. Pediatric Neuroimaging. Fifth ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2012.
- Puppala B, Mangurten HH, McFadden J, et al. Nasal glioma. Presenting as neonatal respiratory distress. Definition of the tumor mass by MRI. Clin Pediatr. 1990;29:49-52.
- Chen CS, David D, Hanieh A. Morning glory syndrome and basal encephalocele. Childs Nerv Syst. 2004;20:87-90.
- Hedlund G. Congenital frontonasal masses: Developmental anatomy, malformations, and MR imaging. Pediatr Radiol. 2006;36:647-662; quiz 726-647.
- Hong SH, Chung HW, Choi JY, et al. MRI findings of subcutaneous epidermal cysts: Emphasis on the presence of rupture. AJR Am J Roentgenol. 2006;186:961-966.
- Ikushima I, Korogi Y, Makita O, et al. MR imaging of Tornwaldt’s cysts. AJR Am J Roentgenol. 1999;172:1663-1665.
- Katzman GL, Dagher AP, Patronas NJ. Incidental findings on brain magnetic resonance imaging from 1000 asymptomatic volunteers. JAMA. 1999;282:36-39.
- Hollender AR. The nasopharynx: A study of 140 autopsy specimens. Laryngoscope. 1946;56: 282-304.
- Maran AG, Buchanan DR. Branchial cysts, sinuses and fistulae. Clin Otolaryngol Allied Sci. 1978;3:77-92.
- Benson MT, Dalen K, Mancuso AA, et al. Congenital anomalies of the branchial apparatus: Embryology and pathologic anatomy. Radiographics. 1992;12:943-960.
- Mancuso AA. Head and Neck Radiology. Vol 2. 2 ed. Philadelphia: Lippincott Williams and Wilkins; 2011.
- Ibrahim M, Hammoud K, Maheshwari M, Pandya A. Congenital cystic lesions of the head and neck. Neuroimaging Clin N Am. 2011;2:621-639, viii.
- Lev S, Lev MH. Imaging of cystic lesions. Radiol Clin North Am. 2000;38:1013-1027.
- Telander RL, Deane SA. Thyroglossal and branchial cleft cysts and sinuses. Surg Clin North Am. 1977;57:779-791.
- Koeller KK, Alamo L, Adair CF, Smirniotopoulos JG. Congenital cystic masses of the neck: Radiologic-pathologic correlation. Radiographics. 1999;19:121-146; quiz 152-123.
- Gutierrez C, Bardaji C, Bento L, et al. Branchio-oto-renal syndrome: Incidence in three generations of a family. J Pediatr Surg. 1993;28: 1527-1529.
- Mazzara CA, Respler DS, Jahn AF. Neonatal respiratory distress: Sequela of bilateral nasolacrimal duct obstruction. Int J Pediatr Otorhinolaryngol. 1993;25:209-216.
- Yousem DM. Neuroradiology. 3rd ed. Philadelphia: Mosby; 2010.
- Bappal A SS, Rathnakar U P, Udupa A L, Prakash S. Conservative management of congenital dacrocystocele. Journal of Clinical and Diagnostic Research. 2010 15 2010;4:2531-2532.
- Wong RK, VanderVeen DK. Presentation and management of congenital dacryocystocele. Pediatrics. 2008;122:e1108-1112.
- Miyagawa Y, Nakazawa M, Kudoh T. Epidermal nevus syndrome associated with anterior scleral staphyloma and ectopic bone and cartilaginous intraocular tissue. Jpn J Ophthalmol. 2010;54:15-18.
- Reddy MA, Francis PJ, Berry V, et al. A clinical and molecular genetic study of a rare dominantly inherited syndrome (MRCS) comprising of microcornea, rod-cone dystrophy, cataract, and posterior staphyloma. Br J Ophthalmol. 2003;87:197-202.
- Agrawal PK, Kumar H. Microphthalmos with cyst: A clinical study. Indian J Ophthalmol. 1993;41:177-179.
- Waring GO, 3rd, Roth AM, Rodrigues MM. Clinicopathologic correlation of microphthalmos with cyst. Am J Ophthalmol. 1976;82:714-721.
- Gujar SK, Gandhi D. Congenital malformations of the orbit. Neuroimaging Clin N Am. 2011;21:585-602, viii.
- Ho CK, Kaufman RL, Podos SM. Ocular colobomata, cardiac defect, and other anomalies: A study of seven cases including two sibs. J Med Genet. 1975;12:289-293.
- Pagon RA. Ocular coloboma. Surv Ophthalmol.1981;25:223-236.
- al-Dousary S. Current management of thyroglossal-duct remnant. J Otolaryngol. 1997;26:259-265.
- Ahuja AT, King AD, Metreweli C. Sonographic evaluation of thyroglossal duct cysts in children. Clin Radiol. 2000;55:770-774.
- Allard RH. The thyroglossal cyst. Head Neck Surg. 1982;5:134-146.
- el-Silimy OE, Bradley PJ. Thyroglossal tract anomalies. Clin Otolaryngol Allied Sci. 1985;10:329-334.
- King AD, Ahuja AT, Mok CO, Metreweli C. MR imaging of thyroglossal duct cysts in adults. Clin Radiol. 1999;54:304-308.
- Branstetter BF, Weissman JL, Kennedy TL, Whitaker M. The CT appearance of thyroglossal duct carcinoma. AJNR Am J Neuroradiol. 2000;21:1547-1550.
- Burnett JW, Goldner R, Calton GJ. Cowden disease. Report of two additional cases. Br J Dermatol. 1975;93:329-336.
- Bajaj Y, Ifeacho S, Tweedie D, et al. Branchial anomalies in children. Int J Pediatr Otorhinolaryngol. 2011;75:1020-1023.
- Harnsberger HR, Mancuso AA, Muraki AS, et al. Branchial cleft anomalies and their mimics: Computed tomographic evaluation. Radiology. 1984;152:739-748.
- Bailey H. Branchial cysts and other essays on surgical subjects in the facio-cervical region. London: Lewis; 1929.
- Raveh E, Papsin BC, Forte V. Branchio-oculo-facial syndrome. Int J Pediatr Otorhinolaryngol. 30 2000;53:149-156.
- Foley DS, Fallat ME. Thyroglossal duct and other congenital midline cervical anomalies. Semin Pediatr Surg. 2006;15:70-75.
- Van Epps KJ, Kuszyk BS, Hofmann LV, Fishman EK. Epidermoid inclusion cysts seen on CT of a patient with Gardner’s syndrome. AJR Am J Roentgenol. 1999;173:858-859.
- Hunter TB, Paplanus SH, Chernin MM, Coulthard SW. Dermoid cyst of the floor of the mouth: CT appearance. AJR Am J Roentgenol. 1983;141:1239-1240.
- Parker GD, Harnsberger HR, Smoker WR. The anterior and posterior cervical spaces. Semin Ultrasound CT MR. 1991;12:257-273.
- Joshi MJ, Provenzano MJ, Smith RJ, et al. The rare third branchial cleft cyst. AJNR Am J Neuroradiol. 2009;30:1804-1806.
- Rea PA, Hartley BE, Bailey CM. Third and fourth branchial pouch anomalies. J Laryngol Otol. 2004;118:19-24.
- Jeyakumar A, Hengerer AS. Various presentations of fourth branchial pouch anomalies. Ear Nose Throat J. 2004;83:640-642, 644.
- Nicollas R, Guelfucci B, Roman S, Triglia JM. Congenital cysts and fistulas of the neck. Int J Pediatr Otorhinolaryngol. 2000;55:117-124.
- Nicoucar K, Giger R, Pope HG, Jr., et al. Management of congenital fourth branchial arch anomalies: A review and analysis of published cases. J Pediatr Surg. 2009;44:1432-1439.
- Franciosi JP, Sell LL, Conley SF, Bolender DL. Pyriform sinus malformations: A cadaveric representation. J Pediatr Surg. 2002;37: 533-538.
- Amanatidou V, Mavrokosta M, Kourtesis A, et al. Cervicomediastinal thymic cyst--report of a case. Thorac Cardiovasc Surg. 2008;56: 177-178.
- Nguyen Q, deTar M, Wells W, Crockett D. Cervical thymic cyst: Case reports and review of the literature. Laryngoscope. 1996;106:247-252.
- 7Cigliano B, Baltogiannis N, De Marco M, et al. Cervical thymic cysts. Pediatr Surg Int. 2007;23:1219-1225.
- Yasufuku M, Maeda K, Takano Y. Thymopharyngeal duct cyst: An unusual cause of respiratory compromise. Pediatr Surg Int. 2009;25: 807-809.
- Hendrickson M, Azarow K, Ein S, et al. Congenital thymic cysts in children—mostly misdiagnosed. J Pediatr Surg. 1998;33:821-825.
- Kondo K, Miyoshi T, Sakiyama S, et al. Multilocular thymic cyst associated with Sjogren’s syndrome. Ann Thorac Surg. 2001;72:1367-1369.
- Smith L, Hamilton JS. Pediatric submental cavernous lymphangioma. Ear Nose Throat J. 2006;85:372-373.
- Fung K, Poenaru D, Soboleski DA, Kamal IM. Impact of magnetic resonance imaging on the surgical management of cystic hygromas. J Pediatr Surg. 1998;33:839-841.
- Bill AH, Jr., Sumner DS. A unified concept of lymphangioma and cystic hygroma. Surg Gynecol Obstet. 1965;120:79-86.
- Liu ME, Branstetter BFt, Whetstone J, Escott EJ. Normal CT appearance of the distal thoracic duct. AJR Am J Roentgenol. 2006;187: 1615-1620.
- Wax MK, Treloar ME. Thoracic duct cyst: An unusual supraclavicular mass. Head Neck. 1992;14:502-505.
- Hekiert A, Newman J, Sargent R, Weinstein G. Spontaneous cervical lymphocele. Head Neck. 2007;29:77-80.
- Wang YA, Zhang ZY, Zheng JW, et al. Spontaneous thoracic duct cyst presenting as a left supraclavicular mass - report of a case and review of literature. Phlebology. 2009;24:82-84.
- Brauchle RW, Risin SA, Ghorbani RP, Pereira KD. Cervical thoracic duct cysts: A case report and review of the literature. Arch Otolaryngol Head Neck Surg. 2003;129:581-583.