Aceruloplasminemia
Aceruloplasminemia
Findings
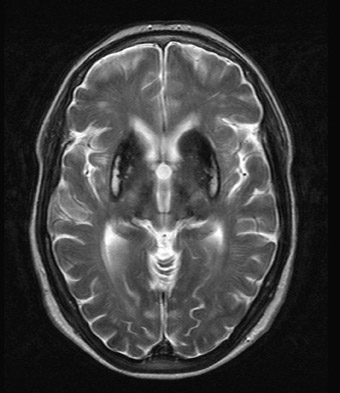
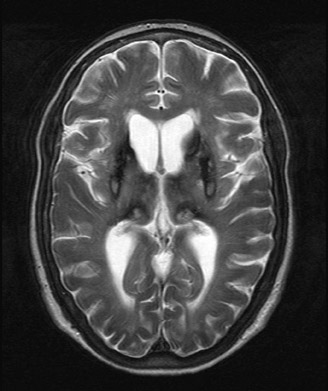
An MRI study revealed marked T2 hypointensity consistent with susceptibility effect involving the putamen, head of the caudate, posterior thalami, and dentate nuclei compatible with iron deposition. In addition, some minimal iron deposition was detected within the cerebral and cerebellar cortices.
Discussion
Iron plays a crucial role in the formation of toxic oxygen radicals. Excess iron functions as a potent catalyst of biologic oxidation. Homeostatic mechanisms are utilized to maintain appropriate serum iron concentrations, thus preventing neuronal cell damage from ironmediated free radicals. Several hereditary disorders, including aceruloplasminemia, can disrupt this homeostasis. Ceruloplasmin is an α2glycoprotein synthesized in hepatocytes that binds plasma copper and has ferroxidase activity.
Aceruloplasminemia is an autosomal recessive disorder characterized by a lack of ceruloplasmin ferroxidase activity important for iron conversion and exportation. The lack of ceruloplasmin results in the accumulation of ferrous iron leading to parenchymal and reticuloendothelial iron overload. Missense mutations in the ceruloplasmin gene (Cp), chromosome 3q, are associated with aceruloplasminemia. Clinically, patients present with a triad of adult onset neurodegeneration, retinal degeneration, and diabetes mellitus.1–3 Laboratory analysis, which is used to confirm the diagnosis, reveals elevated serum ferritin with low serum iron, low transferrin saturation and mild anemia. In addition, ceruloplasmin ferroxidase activity is completely lacking.4
Neurological symptoms include ataxia, dementia, and involuntary movements, thus reflecting the sites of iron deposition.5 The unique involvement of the central nervous system (CNS) distinguishes aceruloplasminemia from other iron or paramagnetic element storage disorders including hemochromatosis, HallervordenSpatz, and Wilson’s disease.6 Autopsy series in aceruloplasminemia have demonstrated severe iron deposition and extensive neuronal loss within the neostriatum, dentate nucleus, and thalamus and marked loss of Perkinje cells within the cerebellar cortex.7,8 MRI demonstrates marked T2 hypointensity within the basal ganglia, with greatest involvement of the putamina and caudate nuclei. T2 hypointensity has also been reported in the thalamic and dentate nuclei.2
In contradiction, hemochromatosis preferentially deposits iron in the pituitary gland. The basal ganglia are typically normal in hemochromatosis. Hallervorden-Spatz syndrome is characterized by iron deposition in the globus pallidus, red nuclei and substantia nigra. MRI demonstrates T2 hypointensity within the basal ganglia with a characteristic T2 high signal in the globus pallidus often referred to as the “eye of the tiger” sign. Additionally, cortical and caudate atrophy can be identified. Wilson’s disease is characterized by the deposition of copper in the basal ganglia and thalami characterized by T2 hypointensity in the lentiform nuclei and thalami. However, a peripheral rim of T2 hyperintensity may surround the lentiform nuclei and ventral mass of the thalami.2
Conclusion
Aceruloplasminemia is an autosomal recessive disorder characterized by abnormal deposition of iron in the brain, resulting in progressive neurological dysfunction. Abnormal deposition of iron and/or other paramagnetic elements in the CNS also occurs in other hereditary disorders such as hemochromatosis, Hallervorden-Spatz syndrome, and Wilson’s disease. Aceruloplasminemia has unique involvement of the CNS compared with the aforementioned disorders that can be demonstrated with MRI.
- Bosio S, De Gobbi M, Roetto A, et al. Anemia and iron overload due to compound heterozygosity for novel ceruloplasmin mutations. Blood. 2002;100:22462248.
- Grisoli M, Piperno A, Chiapparini L, et al. MR imaging of cerebral cortical involvement in aceruloplasminemia. AJNR Am J Neuradiol. 2005;26: 657661.
- Ponka P. Hereditary causes of disturbed iron homeostasis in the central nervous system. Ann of the New York Academy of Sciences. 2004;1012: 267281.
- Xu X, Pin S, Gathinji M, et al. Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Annals of the New York Academy of Science.s 2004;1012:299305.
- Miyajima H, Takahashi Y, Kono S. Aceruloplasminemia, an inherited disorder of iron metabolism. Biometals. 2003;16:205213.
- Miyajima H, Kono S, Takahashi Y, Sugimoto M. Increased lipid peroxidation and mitochondrial dysfunction in aceruloplasminemia brains. Blood, Cells, Molecules and Diseases 2002;29:433438.
- Kaneko K, Yoshida K, Arima K, et al. Astrocytic deformity and globular structures are characteristic of the brain of patients with aceruloplasminemia. Journal of Neuropathology and Experimental Neurology 2002;61:10691077.
- Miyajima H. Aceruloplasminemia, an iron metabolic disorder. Neuropathology 2003;23:345350.